



Abstract
Glioblastoma, the most aggressive cerebral tumor, is invariably lethal. Glioblastoma cells express several genes typical of normal neural stem cells. One of them, SOX2, is a master gene involved in sustaining self-renewal of several stem cells, in particular neural stem cells. To investigate its role in the aberrant growth of glioblastoma, we silenced SOX2 in freshly derived glioblastoma tumor-initiating cells (TICs). Our results indicate that SOX2 silenced glioblastoma TICs, despite the many mutations they have accumulated, stop proliferating and lose tumorigenicity in immunodeficient mice. SOX2 is then also fundamental for maintenance of the self-renewal capacity of neural stem cells when they have acquired cancer properties. SOX2, or its immediate downstream effectors, would then be an ideal target for glioblastoma therapy.
Introduction
Cerebral tumors account for about 2% of all cancers, with an incidence rate of about 14 new cases per 100,000 individuals each year [1]. Gliomas are the most frequent cerebral neoplasias (86%) and are clinically divided into four stages, glioblastoma (World Health Organization grade IV) being the most common and aggressive [2]. Despite its relatively low frequency, this tumor is responsible for 4% of all deaths caused by cancer, because, notwithstanding combined therapy, the median survival duration is 14 months with very few long-term survivors. One of the reasons for this poor prognosis is the inherent complexity of the tumor. Glioblastoma cells deeply infiltrate the surrounding tissue in a way highly reminiscent of the ability to migrate to long distances typical of neural stem cells. Consistent with the hypothesis that this tumor originates from neural stem cells, glioma cells express a number of molecules associated with the development of normal neural stem cells [3, 4]. One of them, the transcription factor SOX2, appears especially interesting due to its role in sustaining growth and self-renewal of several stem cell types, both embryonic and adult [5–9]. In conjunction with Oct3/4 and Nanog, SOX2 is considered a master gene of mammalian embryogenesis and may be part of a complex network of transcription factors that affect both pluripotency and differentiation in embryonic stem cells [8]. In the mouse nervous system SOX2 is expressed in neural stem cells, early precursors, and a few mature neurons, and is essential in maintaining cell proliferative potential, as shown by several experiments in which the transcription factor was either overexpressed or knocked down [10, 11]. These experiments also show that the effect of SOX2 is clearly dosage-dependent [12].
SOX2 has been found to be expressed in a variable percentage of cells in several malignant tissues [13–17]. In a recent paper, SOX2 was found to be expressed in all glioma samples tested [13]. In agreement with the heterogeneity of the cell population present in each glioma, typically containing cells at various stages of differentiation, SOX2 is expressed in a highly variable percentage of cells, in the range of 6%–80% in different samples ([13] and our own data).
These data and the known role of SOX2 in development and cell differentiation suggest that this transcription factor may be relevant to the aberrant growth of glioma cells.
We thus decided to knock down the expression of SOX2 in glioma cells using retroviral vectors harboring a microRNA (miRNA) engineered to target SOX2 mRNA. We chose to use cells freshly derived from tumor samples kept in culture under the conditions used for neural stem cell growth. These cultures are able to grow indefinitely both as neurospheres and adhered to Matrigel, and when injected orthotopically in immunodeficient mice, produce a tumor histologically very similar to the original tumor. As recently shown, glioma cells cultured under these conditions are a far better model of human glioblastoma than the established cell lines used so far [18]. The term tumor-initiating cells (TICs) is frequently used to describe cells such as these with cancer stem cell capacities [19], and we shall then use this term throughout this paper.
Our results indicate that SOX2 is indeed necessary for the continuous proliferation of human glioma cells, both in vitro and in the brain of immunodeficient mice.
Materials and Methods
Human Tumors and Glioma TIC Culture
Tumor samples were obtained during surgery from patients of the Neurosurgery Department, San Martino Hospital (Genova, Italy). Informed consent was obtained for all patients before the surgery as approved by the ethics board.
Samples were collected immediately after surgical resection and processed for isolation of TICs, according to Lee et al. with modifications. Briefly, the tissue was washed, minced, and triturated, and the cells were seeded at a concentration of 100,000 per ml into tissue culture flasks coated with Matrigel (BD Biosciences, San Diego, http://www.bdbiosciences.com). Cells were grown in proliferation medium containing Dulbecco's modified Eagle's medium (DMEM)-F12/Neurobasal and B27 supplement, 2 mM L-glutamine (Gibco-Invitrogen, Grand Island, NY, http://www.invitrogen.com), recombinant human fibroblast growth factor-2 (10 ng/ml; Peprotech, Rocky Hill, NJ, http://www.peprotech.com), and recombinant human epidermal growth factor (EGF) (20 ng/ml; Peprotech). The proliferation medium was changed twice a week. Under these conditions, the cells attach and grow as a monolayer in flasks and maintain intact self-renewal capacity.
Differentiation Assay
To induce differentiation, single-cell suspensions of glioblastoma TICs were seeded on Matrigel-coated coverslips at 1 × 104 cells per slide and cultured for up to 21 days under differentiation conditions in DMEM-F12/Neurobasal containing B27 supplement (2% v/v), 2 mM L-glutamine, 100 U/ml penicillin/streptomycin (all Gibco-BRL, Gaithersburg, MD, http://www.gibcobrl.com) and fetal calf serum (FCS) (10% v/v; Euroclone, Siziano, Italy, http://www.euroclonegroup.it).
Intracranial Tumor Assays
Tumorigenicity in nonobese diabetic severe combined immunodeficient (NOD–SCID) mice was used to check that the glioma cell cultures maintained brain tumor-initiating activity. The animals were housed in pathogen-free conditions, and the review board of the Istituto Nazionale per la Ricerca sul Cancro approved the experiments, performed according to the National Regulation on Animal Research Resources. For intracranial inoculation, two to four NOD–SCID mice (6–8 weeks old; Charles River Laboratories International, Inc., Wilmington, MA, http://www.criver.com), for each experimental condition, were anesthetized with i.m. ketamine and xylazine [20]. Thereafter, the animal was positioned into a stereotactic frame (Model 900; David Kopf Instruments, Tujunga, CA, http://www.kopfinstruments.com) and a hole was made using a 21-gauge needle, 2 mm lateral and 1 mm anterior from the intersection of the coronal and sagittal sutures (bregma). Cells (105) were injected using a Hamilton syringe (series 7000; Sigma-Aldrich, St. Louis, http://www.sigmaaldrich.com) at a depth of 3.5 mm in a volume of 2 μl in the left corpus striatum. Mice were monitored for 6 months for disease symptoms and were sacrificed by CO2 asphyxiation when they showed weight loss or any other sign of disease ranging from hyper-reactivity to imbalanced stance and gait. All experiments including animals were performed in compliance with guidelines approved by the ethical committee for animal use in cancer research at Istituto Nazionale Ricerca sul Cancro–Genova.
Generation of the Antisera Against SOX2 and BF1
The antisera were raised in New Zealand rabbits using as immunogen the C-terminal domain of human SOX2 and human BF1. The specificity of the antisera was tested in several cell lines by immunocytochemistry and Western blotting.
Immunocytochemistry
Immunocytochemistry was performed to examine the expression of neural stem cell markers and lineage markers. For immunostaining, the cells plated on Matrigel-coated glass coverslips were fixed with 4% paraformaldehyde (PFA) for 15 minutes at room temperature, treated with phosphate-buffered saline (PBS), 10% FCS, and 0.3% Triton X100, and then stained with the following antibodies: anti-Nestin (mouse monoclonal, 1:1000; Novus Biologicals, Littleton, CO, http://www.novusbio.com), anti-Map2 (rabbit polyclonal, 1:1,000; Chemicon, Temecula, CA, http://www.chemicon.com), anti-glial fibrillary acidic protein (GFAP) (rabbit polyclonal, 1:10,000; DAKO, Glostrup, Denmark, http://www.dako.com), anti-Ki67 (mouse monoclonal, 1:700; Oncogene, Cambridge, MA, http://www.oncogene.com), chicken polyclonal anti-green fluorescent protein (GFP) (1:500; Abcam, Cambridge, U.K., http://www.abcam.com), anti-SOX2 (rabbit polyclonal, 1:500; Chemicon and anti-SOX2 rabbit polyclonal produced in our laboratory, 1:500), anti-Olig2 (rabbit polyclonal, 1:100; Chemicon), anti-O4 (mouse monoclonal, 1:200: Chemicon), anti-Emx2 [21], and anti-BF1 (rabbit polyclonal produced in our laboratory, 1:1,000). The primary antibodies were detected with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (1:700; Alexa, Gibco-Invitrogen), rhodamine-conjugated goat anti-rabbit IgG (1:250; Jackson ImmunoResearch Laboratories, West Grove, PA, http://www.jacksonimmuno.com), and FITC-conjugated goat polyclonal anti-chicken IgY (1:400; Abcam). The cells were counterstained with Hoechst 33342 dye (Sigma) to identify all nuclei. Quantification of cells positive for a specific marker was carried out by counting all the stained cells within 20 randomly selected microscope fields per specimen, and the percentage was calculated based on the total number of nuclei counted.
Immunohistochemistry
For xenograft tumor analysis, animals were sacrificed, brains were cryopreserved, and 10-mm cryostat (CM 1100; Leica Microsystems GmbH, Wetzlar, Germany, http://www.leica-microsystems.com) sections were cut. Sections bearing tumors were identified by hematoxylin and eosin staining. Briefly, brain sections were mounted on slides and first stained with Harris hematoxylin for 40 seconds, and then counterstained with alcoholic eosin for 30 seconds. Cryosections containing tumors were permeabilized in PBS containing 0.2% Triton X-100 and blocked in 5% normal FCS-PBS. After incubation with primary antibodies in FCS-PBS overnight at 4°C, sections were washed in tris-buffered saline, stained with the appropriate secondary antibodies, and counterstained with Hoechst 33342 dye to identify all nuclei. Images were obtained with a Nikon Eclipse 80i microscope (Nikon Instruments, Inc., Melville, NY, http://www.nikoninstruments.com).
Vector Construction
miRNA Vectors.
Oligonucleotides targeting a human SOX2 sequence compatible for use in cloning into BLOCK-iT Pol II miR RNAi expression vectors were obtained using the online tool BLOCK-iT RNAi Designer.
1041: top strand, 5′-TGCTGTGGTCATGGAGTTGTACTGCAGTTTTGGCCACTGACTGACTGCAGTACCTCCATGACCA-3′; bottom strand, 5′-CCTGTGGTCATGGAGGTACTGCAGTCAGTCAGTGGCCAAAACTGCAGTACAACTCCATGACCAC-3′.
1238: top strand, 5′-TGCTGATACATGCTGATCATGTCCCGGTTTTGGCCACTGACTGACCGGGACATTCAGCATGTAT-3′; bottom strand, 5′-CCTGATACATGCTGAATGTCCCGGTCAGTCAGTGGCCAAAACCGGGACATGATCAGCATGTATC-3′.
3′-untranslated region (UTR): top strand, 5′-TGCTGTTCTCCATGCTGTTTCTTACTGTTTTGGCCACTGACTGACAGTAAGAAAGCATGGAGAA-3′; bottom strand, 5′-CCTGTTCTCCATGCTTTCTTACTGTCAGTCAGTGGCCAAAACAGTAAGAAAC- AGCATGGAGAAC-3′.
The pcDNA 6.2-GW/EmGFP-miR-neg control plasmid contains an insert that can form a hairpin structure that is processed into mature miRNA, but is predicted not to target any known vertebrate gene. The negative control sequence without 5′ overhangs is shown below:
GAAATGTACTGCGCGTGGAGACGTTTTGGCCACTGACTGACGTCTCCACGCAGTACATTT
Cloning procedures were performed following the manufacturer's instructions.
The LXSN retroviral vector (Clontech, Palo Alto, CA, http://www.clontech.com) was constructed as a destination vector following the Gateway Vector Conversion System (Invitrogen) after excision of the simian virus 40 promoter and neo sequence.
L-SOX2-IN Retroviral Vector.
The coding sequence of the human SOX2 gene was cloned in LXIN retroviral vector (Clontech).
Generation of Viruses
A retrovirus was generated by transfection of the Phoenix 293T amphotropic packaging cell line with miRNA–short-hairpin RNA (shRNA)-expressing retroviral vectors or with the L-SOX2-IN retroviral vector. Briefly, Phoenix cells were cultured at 37°C, 5% CO2, in 100-mm tissue culture dishes (Corning Life Sciences, Acton, MA, http://www.corning.com/lifesciences) using DMEM (Gibco-Invitrogen), 10% FBS (Gemini Bio-Products, West Sacramento, CA, http://www.gembio.com), 2 mM glutamine (Gibco-Invitrogen), and 100 U/ml penicillin with 100 μg/ml streptomycin (Gibco-Invitrogen). Ten micrograms per dish of retroviral vector diluted in Opti-MEM (Gibco-Invitrogen) was transfected using Lipofectamine 2000 reagent (Invitrogen). At around 16 hours post-transfection, the medium on the Phoenix cells was replaced with nondifferentiating medium. The viral supernatant was harvested at 48 hours post-transfection, spun at 1,200 rpm for 5 minutes, and filtered through a 0.45-μm filter. Polybrene was added to a final concentration of 8 μg/ml. The medium was replaced at around 16 hours postinfection. Infection was repeated four times. Cells infected with L-SOX2-IN retrovirus were cultured for 1 week in 1 mg/ml of G418 for selection.
Cloning Assay
Five hundred or one thousand cells were plated on Matrigel-coated coverslips 4 days or 4 weeks after infection. Cells were fixed with 4% PFA for 10 minutes and stained with different antibodies after 1 and 2 weeks. The number of cells per clone was counted by two different investigators, scoring at least 500 clones.
Separation of GFP+ Cells by Fluorescence-Activated Cell Sorting
Four days after the last infection, cells were trypsinized and resuspended in PBS at 106 cells/ml. Cells were sorted using a fluorescence-activated cell sorting (FACS) Aria Cell Sorter (BD Biosciences) focusing to the highest possible purity of GFP+ cells.
Murine Neural Precursor Production and Culture
Neural precursors were derived from the periventricular region of E18.5 CD1 embryos as previously described [21–23].
Data Analysis
TIC cultures were performed in at least triplicate. Data were compared using the Student's t-test, and a p-value <.05 was considered statistically significant.
Results
In Vitro Characterization of TICs Derived from Adult Human Glioblastoma
Cells dissociated from biopsies or surgical samples were plated in serum-free medium in the presence of EGF and basic fibroblast growth factor. One to two weeks after plating, cellular aggregates resembling neurospheres derived from normal neural precursors were detectable in all glioblastoma cultures. Seven of 12 cultures established from glioblastomas gave rise to long-term expanding cultures. Cells were cultured either as spheres or as a monolayer adhered to Matrigel.
The glioblastoma cells could be exponentially expanded, with a doubling time that varied in the range of 3–7 days in cultures derived from different patients. To assess the differentiation ability of our glioblastoma-derived cells, we tested their ability to differentiate upon removal of growth factors, adhesion to Matrigel, and addition of serum. The expressions of Nestin and SOX2, known to be present both on normal and glioblastoma precursors, were assessed in nondifferentiating and differentiating conditions. Expressions of Ki67, GFAP, and Map2 were also analyzed as markers of proliferation, glial differentiation, and neuronal differentiation, respectively. Table 1 summarizes the results obtained with TICs derived from seven patients. Under nondifferentiating conditions, most of the cells were positive for Nestin and SOX2 in all TICs, whereas only three of them expressed high levels of a differentiation marker, Map2 in patients F and L and GFAP in patient D. The vast majority of the Ki67+ cells were concentrated in the SOX2+ fraction. When cultured in differentiating medium, all TICs showed some degree of differentiation, although this varied among the different patients. Thus, the proliferation marker Ki67 drastically decreased in all patients, whereas in patients C, P, and L Nestin decreased only partially. The percentage of Map2+ and GFAP+ cells increased in all patients, although, unlike normal neural precursors, a fraction of the differentiated cells coexpressed glial and neuronal markers, as previously described for glioblastoma stem cells [4, 24]. SOX2 decreased in all TICs except in patient V, maintaining a strong association with Ki67.
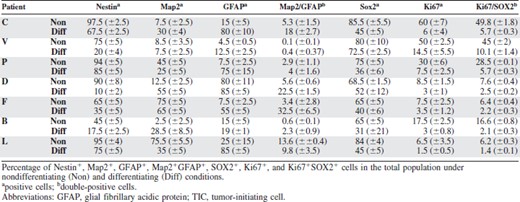
These findings show that the glioblastoma cultures contain a population capable of differentiation and long-term self-renewal, as expected for TICs.
We then assessed their ability to initiate tumors in vivo when orthotopically injected in immunodeficient mice.
Tumorigenic Properties of Human Glioblastoma Precursors
Orthotopic transplantation of as few as 5,000 TICs, in immunodeficient NOD–SCID mice, reproducibly generated serially transplantable, highly infiltrating tumors with features similar to the original glioblastoma (supporting information Fig. 1). In particular, one of them, patient V, was able to infiltrate the host brain almost completely, except for the cerebellum. Mice were sacrificed at the first appearance of neurological symptoms, ranging from hyper-reactivity to imbalanced stance and gait (75–90 days after transplantation), and frozen sections of the brains were stained with antibodies specific for SOX2 (Fig. 6K), and for human Nestin and Ki67 (supporting information Fig. 1). A high percentage of cells was immunoreactive for all three markers in all tumors.
Silencing of SOX2 in Glioblastoma Cells
To investigate the function of SOX2 in glioblastoma cells we silenced SOX2 gene expression in the seven TICs kept under nondifferentiating conditions. To achieve stable knockdown of SOX2 in these cells, we cloned shRNA oligonucleotides directed against human SOX2 within a mir155 sequence into the PLX retroviral vector, which contains GFP as a reporter. Three different miRNAs were designed to silence SOX2 targeting different sequences of the coding region of SOX2 mRNA (see Materials and Methods). One of them (1041) gave a nearly complete knockdown of a SOX2 transgene following cotransfection into the 293T cell line (Fig. 1, lane 4) and was therefore used for all subsequent experiments. The same vector containing an irrelevant miRNA sequence was used as a control.
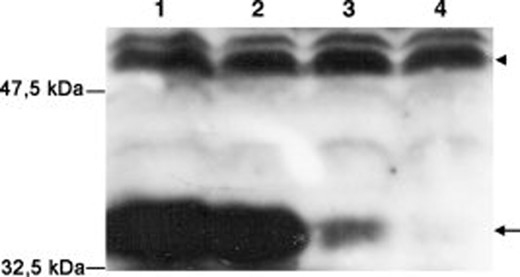
Silencing efficiency of miRNA targeting different SOX2 sequences. Western blot analysis of SOX2 expression (arrow) in 293T cells cotransfected with a SOX2 expression vector (all lanes) and silencing retroviral vectors containing: lane 1, GFP only; lane 2, control sequence; lane 3, 3′-UTR sequence; lane 4, sequence 1041. Arrow indicates SOX2. Arrowhead indicates α-tubulin. Abbreviations: GFP, green fluorescent protein; miRNA, microRNA; UTR, untranslated region.
The efficiency of infection varied in the seven TICs in the range of 6%–50%, probably reflecting the different proliferation rates of the individual TICs. Only in TICs derived from patients V, C, and P was the level of infection, 50%, 40%, and 20%, respectively, sufficiently high to allow a statistical evaluation of the results, without having to infect an exceedingly high number of cells, and these three patients were therefore used in all subsequent experiments.
Immunostaining of the cells with an anti-SOX2 antiserum 4 days after infection showed that the virus was indeed able to knockdown SOX2 expression in all TICs, although the level of downmodulation varied among them (Figs. 2, 6A–6F). The silencing rate was fairly high, as only about 15% of the GFP+ cells were still SOX2+, probably because they failed to correctly express the silencing miRNA.

SOX2 silencing efficiency in glioblastoma TICs. Percentage of SOX2− cells in GFP+ infected cells, 4 days after infection in patients C, V, and P. Dark bars indicate cells infected with SOX2 silencing vector targeting sequence 1041; gray bars indicate cells infected with control vector. Triplicate samples were used for each group. Error bars indicate mean ± SEM. Abbreviations: GFP, green fluorescent protein; SEM, standard error of the mean; TIC, tumor-initiating cell.
When the transduced cells were cultured under nondifferentiating conditions, the percentage of GFP+ cells decreased slowly but steadily (Fig. 3A), a decline entirely due to the disappearance of the GFP+SOX2− population (Fig. 3B). As shown in Figure 4, this latter population did not completely disappear from the culture, but reached a plateau at about 5% after 5 weeks. However, as stated above, SOX2 had not been silenced in all GFP+ cells, and it is very likely that the residual GFP+SOX2− population is derived from these cells, being very similar to the steady percentage of GFP+SOX2− cells present in the mock transduced TICs (gray line in Fig. 3B).
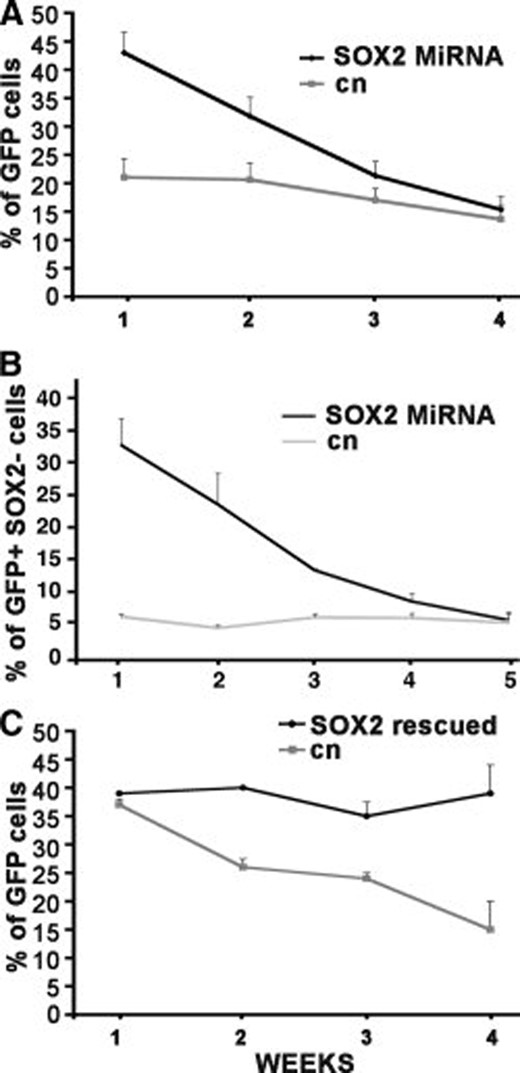
Progressive decrement of SOX2 silenced cells in the total population. (A): Percentage of GFP+ cells measured 1–4 weeks after infection. (B): Percentage of SOX2 silenced cells measured 1–5 weeks after infection. Dark line indicates cells infected with SOX2 silencing vector; gray line indicates cells infected with control vector. Triplicate samples were used for each patient. Error bars indicate mean ± SEM of patients C, V, and P. (C): Percentage of GFP+ cells constitutively expressing SOX2 measured 1–4 weeks after infection. Dark line indicates cells infected with the SOX2 neo vector subsequently infected with the silencing vector directed against the 3′-UTR; gray line indicates cells infected with the control neo vector subsequently infected with the silencing vector directed to the 3′-UTR. See text for details. Patient V, mean of three experiments. Abbreviations: GFP, green fluorescent protein; SEM, standard error of the mean; UTR, untranslated region.
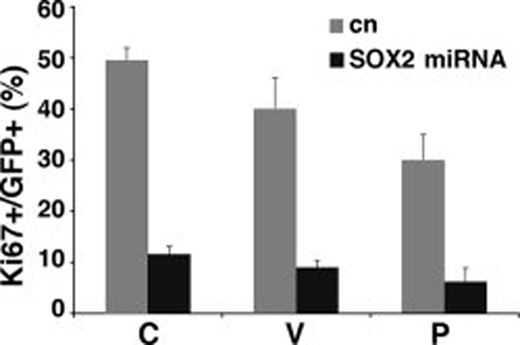
Reduced Ki67 expression in SOX2 silenced cells. Percentage of Ki67+ cells in GFP+ cells 7 days after infection in patients C, V, and P. Dark bars indicate cells infected with SOX2 silencing vector; gray bars indicate cells infected with control vector. Triplicate samples were used for each patient. Error bars indicate mean ± SEM. Abbreviations: GFP, green fluorescent protein; SEM, standard error of the mean.
Accordingly, Ki67 expression was greatly reduced in SOX2 silenced cells. Only a minor fraction of SOX2− cells were positive for this proliferation marker 7 days after SOX2 knockdown (Fig. 4).
To confirm that the observed effect was due to SOX2 knockdown and not to an off-target effect, we infected the cells with the same retroviral vector carrying a different miRNA directed against the 3′-UTR of the SOX2 mRNA. This latter miRNA caused silencing of SOX2 and a reduction in Ki67 expression, similar to the 1041 sequence. Accordingly, the infected glioblastoma cells exited prematurely form the cell cycle and disappeared from the culture with similar kinetics (data not shown). To further confirm the specificity of the observed effect, we performed the following experiment. Cultured TICs from patients C and V were first infected either with a bicistronic retroviral vector carrying a truncated cDNA containing only the coding sequence of SOX2 and the neo resistance gene or with a control vector containing only the neo gene. After 1 week of selection in G418, the cells were then transduced with the silencing vector directed to the 3′-UTR of the SOX2 mRNA. TICs infected with the control vector showed the usual amount of SOX2 silencing. In contrast, nearly all the cells infected with the silencing vector were constitutively expressing SOX2 from the transgene lacking the 3′-UTR, which could not be a target of the silencing vector, and maintained expression of the transcription factor. If the disappearance of the SOX2 silenced cells was due to an off-target effect, then the GFP+ population should be diluted off at the same rate in both cultures. As shown in Figure 3C for TICs derived from patient V, this was not the case. The mock transduced GFP+ cells showed the usual decline, whereas the SOX2 transduced GFP+ cells did not show any significant decrease. The same results were obtained with TICs derived from patient C (not shown).
Clonogenicity of SOX2 Silenced Cells
Four days after miRNA transduction, single-cell suspensions were seeded on Matrigel-coated coverslips and allowed to form colonies for 1 and 2 weeks. The resulting clones were fixed and stained with anti-GFP antibodies and the size of each individual clone was analyzed. The number of GFP+ cells per clone was counted, which revealed a significant reduction (p < .001) in clone size in the SOX2 silenced cells compared with control cells already after 1 week (Fig. 5). After 2 weeks, the difference in proliferation rate was even more pronounced (p < .001) (Fig. 5). As these results could indicate a progressive loss of proliferative ability by the silenced cells, we plated TICs 4 weeks after SOX2 knockdown, when the percentage of GFP+ cells was around 15%, and allowed them to form colonies for 1 week. No GFP+SOX2− clones could be obtained in this case, whereas the average size of the GFP+SOX2+ clones did not differ from that of mock transduced cells (data not shown).
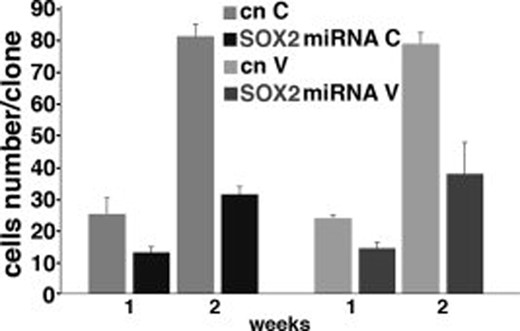
Reduced clone size in SOX2 silenced cells. Clone size of patient C (right) and patient V (left) after 1 and 2 weeks of culture. Dark bars indicate cells infected with SOX2 silencing vector; gray bars indicate cells infected with control vector. Triplicate samples were used in each group. Error bars indicate mean ± SEM. Abbreviation: SEM, standard error of the mean.
Taken together, these observations suggest that the reduced proliferation of SOX2 knockdown cells is not due to a slower cell cycle, but that, in the absence of SOX2, glioblastoma cells have lost the ability to divide indefinitely and are only capable of a limited number of rounds of cell division, after which they exit from the cell cycle.
The figure shows the results for only two patients, because the lower efficiency of transduction did not allow us to obtain an adequately sized sample of clones from the third patient. However, in this case, smaller clones were also obtained after 1 week, which increased only slightly in size in the second week. Again, no GFP+SOX2− clones were obtained when the clonogenicity assay was performed 4 weeks after SOX2 knockdown.
Fate of SOX2 Silenced Cells
To test whether increased programmed cell death or senescence could account for the reduction in cloning efficiency and growth of SOX2 silenced TICs, terminal deoxynucleotidyl transferase dUTP nick end labeling experiments and cytoplasmic β-galactosidase assays were performed without demonstration of significant differences compared with control cells (data not shown).
Given the role of SOX2 in normal cells, these results could be explained by an increased differentiation rate in the SOX2 knockdown population. We therefore evaluated the expression of several markers, Olig2, BF1, Emx2, O4, Nestin, Map2, and GFAP, in the SOX2 silenced cells. A significant change was observed in the expression of Olig2, which decreased from 70% in control TICs to 28% in SOX2 knockdown cells from patient C and decreased from 65% to 36% in patient V, and of BF1, which decreased from 65% in control TICs to 45% in SOX2 knockdown cells from patient C and decreased from 54% to 25% in patient V. The percentage of Emx2 (∼50% in both patients), O4 (∼55% in both patients), and Nestin (∼85% in both patients) positivity in SOX2 silenced cells was not significantly different from that of control cells. In no instance, could we observe a significant increase in the number of cells expressing Map2 or GFAP. However, as shown in Table 1, TIC cultures contain varying proportions of cells expressing Map2 or GFAP, or both, and, under these conditions, it is therefore hard to detect a small increase in the percentage of differentiating cells.
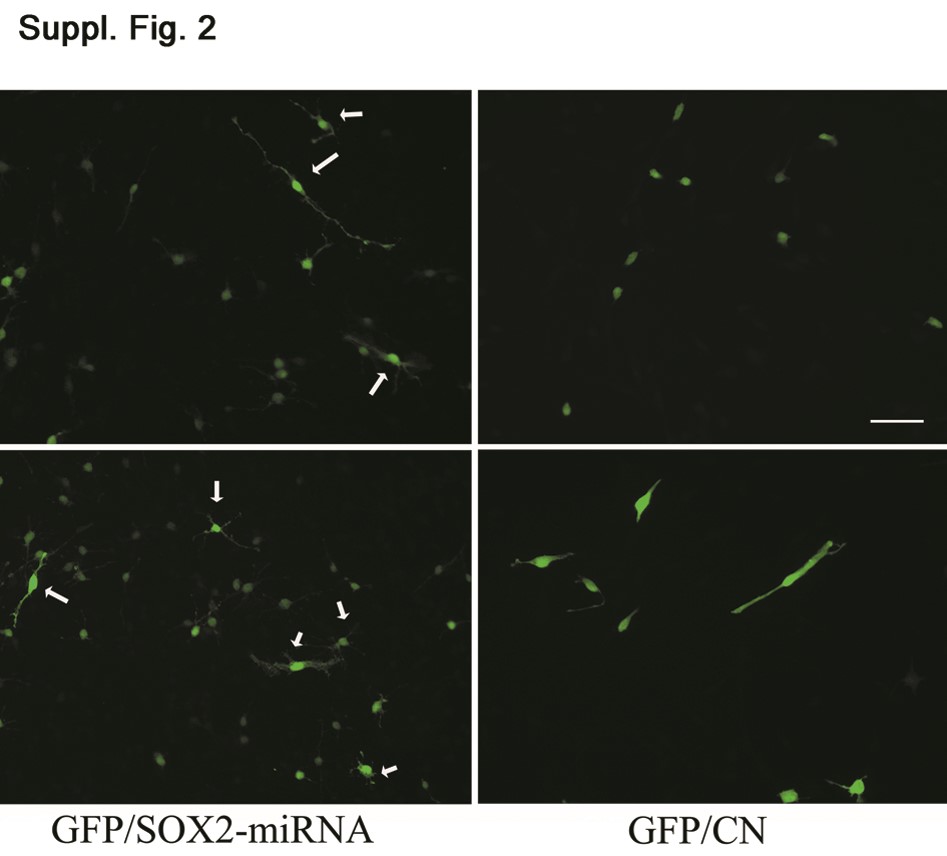
We then performed similar experiments in murine neural precursors, the available closer-to-normal counterpart of glioblastoma TICs, in which small differences in the differentiation rate should be readily detected. The percentage of SOX2+ cells in cultures kept under “stem” conditions was around 90%. SOX2 was then knocked down using the same retroviral vector in E18.5 murine neural precursors kept under nondifferentiating conditions. In this case as well, >80% of GFP+ cells showed SOX2 silencing. Similar to what was observed in the glioblastoma cultures, their proportion decreased with time and was reduced by 50% after 2 weeks. To assess the effect of SOX2 silencing on differentiation, we then stained neural precursors, kept in nondifferentiating conditions, with anti-Map2 and anti-GFAP antibodies 1 week after infection. The percentage of GFP+Map2+ cells was significantly higher (46% ± 5.8%) in SOX2 silenced neural precursors than in cells infected with the control vector (17.7% ± 3.7%; p < .001), in six independent experiments. In addition, most of the GFP+SOX2− cells were elongated and highly branched (supporting information Fig. 2). The low number of GFAP+ cells was not significantly different in both the experimental and control cultures.
We performed a rather high number of experiments because the number of Map2+ cells was unusually high for neurosphere cultures. The infection with the vector itself cannot be responsible for the increase in the number of neuroblasts because uninfected E18.5 murine neural precursors already showed a percentage of Map2+ cells as high as 15.6% ± 2.8%. However, the cultures were kept not as neurospheres but adhered to Matrigel, an essential procedure for subsequent infection. It could then be that these conditions may favor differentiation along the neuroblast lineage.
In Vivo Tumorigenicity of SOX2 Silenced TICs
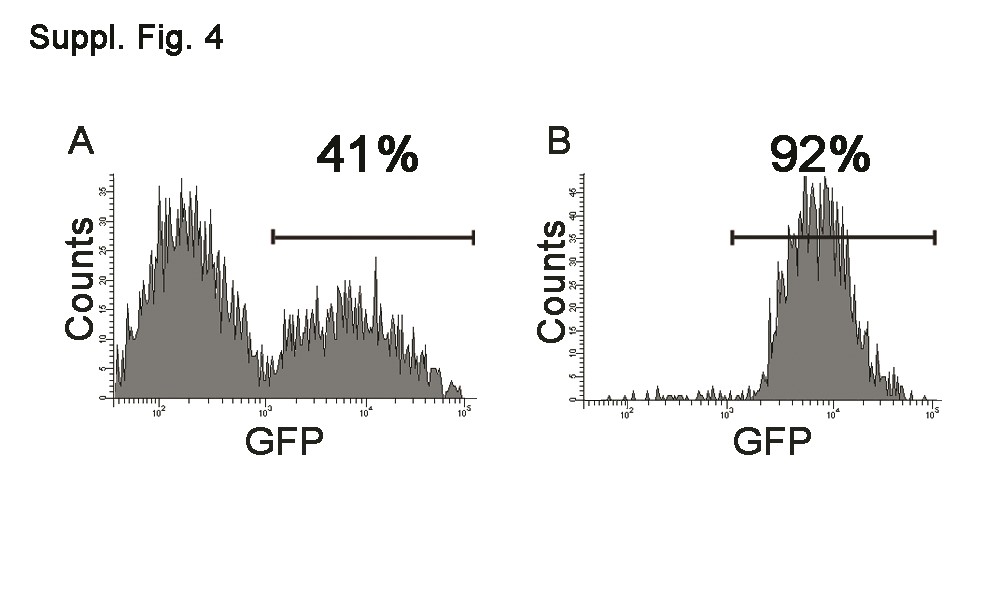
We next tested whether SOX2 silencing affected the tumorigenicity of cultured TICs in NOD–SCID mice. TICs from patients V and C were used because they consistently gave a percentage of infection in the range of 40%–50%. One week after infection with the silencing vector, the rate of infection and silencing were assessed, and 100,000 cells obtained from two independent sets of infections were orthotopically transplanted into NOD–SCID mice. All mice developed tumors at about the same time as animals injected with TICs infected with the control retroviral vector, as expected by the fact that the majority of the transplanted cells were still expressing SOX2. The brains of mice injected with retroviral-infected cells were examined by immunohistochemical and immunofluorescence analysis using SOX2- and GFP-specific antibodies. Distinct tumor masses were detected within the striatum, near the ventricular walls, with cells migrating along the corpus callosum. A great number of transplanted tumor cells showed a significant positivity for SOX2, human-specific Nestin, and Ki67, as observed in uninfected tumors (Fig. 6 and supporting information Fig. 1). Although the proportion of GFP+ cells was lower in the injected control cells than in SOX2 miRNA transduced cells, many tumor masses derived from GFP+ cells were present in the control xenografts (supporting information Fig. 3), demonstrating that cancer stem cells had been infected by the retroviral vector. In contrast, brain masses formed in mice injected with SOX2 miRNA transduced TICs were very rarely GFP+ (supporting information Fig. 3). Moreover, when the sections were double-stained with anti-GFP and anti-SOX2 antibodies, all the tumor areas containing GFP+ cells were also positive for SOX2 (supporting information Fig. 3). To confirm the loss of tumorigenicity by SOX2 silenced TICs, cells derived from patients C and V were infected with the silencing vector and the control vector, and separated according to GFP expression using a FACS sorter. The purity of the sorted fractions in four independent experiments was 80%–95% (see supporting information Fig. 4 for the experiment shown in Fig. 6). GFP+ sorted cells (105) were orthotopically injected into NOD–SCID mice. The animals were sacrificed at the first appearance of neurological signs in the mice injected with the control GFP+ cells, about 3 months after transplantation. As shown in Figure 6 for patient C, whereas the control mice showed a GFP+ tumor mass (Fig. 6J), only a few SOX2+GFP− cells could be detected in the brains of mice injected with the SOX2 silenced cells (Fig. 6G, 6H). In these latter mice, it was also possible to find a few GFP+SOX2+ cells (Fig. 6M, 6N). GFP staining was performed by immunohistochemistry because we found it very hard to spot such cells by immunofluorescence, as they tended to show a lower GFP expression level than the silenced cells.
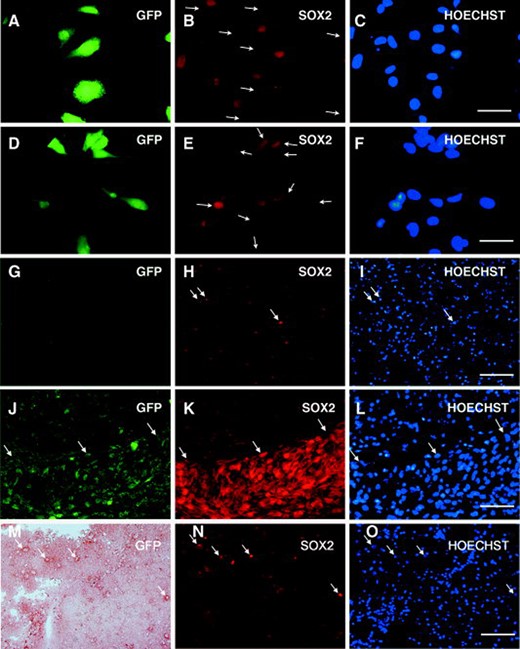
Loss of tumorigenicity by SOX2 silenced cells. Silencing of SOX2 expression in cultured TICs of patient C. (A–C): TICs transduced with the silencing vector. (D–F): TICs transduced with the control vector. Scale bar 12 μm. (G–O): Sections of tumors originated from TICs derived from patient C. (G–I): GFP+ sorted cells from patient C infected with the silencing vector. (J–L): GFP+ sorted cells from patient C infected with the control vector. (M–O): GFP+ sorted cells from patient C infected with the silencing vector. (A, D, G, J, M): GFP staining; (B, E, H, K, N): SOX2 staining; (C, F, I, L, O): Hoechst staining. Arrows in (B) indicate GFP+ SOX2 silenced cells. Arrows in (E) indicate GFP+SOX2+ cells. Arrows in (H, I) indicate SOX2+ cells and the corresponding nuclei. Arrows in (J–L) indicate the border of the tumor mass. Arrows in (M–O) indicate rare GFP+SOX2+ cells and the corresponding nuclei. Scale bar 50 μm. Abbreviations: GFP, green fluorescent protein; TIC, tumor-initiating cell.
Discussion
It is now widely accepted that tumors are mixed populations of cells at various stages of differentiation, only a fraction of which can perpetuate the tumor and are therefore named, in analogy with their normal counterparts, cancer stem cells [19, 24]. Given the impossibility of directly identifying them, due to the lack of suitable markers, these cells are operationally defined as the small fraction of the cancer cells in a malignancy that have the ability to propagate the tumor upon transplantation into immunodeficient mice. The xenotransplanted tumor can be serially transplanted into new recipient mice, highlighting the capacity of perpetual self-renewal of this subset of cancer cells (TICs). We isolated and characterized human glioblastoma TICs from seven patients. These cells, as the tumors from which they derive ([18] and our own data), share genetic and phenotypic features with normal neural stem cells, such as expression of the transcription factor SOX2. The SOX2 gene has been shown to have an essential role in the maintenance of the proliferative potential of neural stem/precursor cells and in ensuring the production of sufficient cell numbers of the appropriate type [10, 12, 25]. SOX2 expression in glioblastoma TICs parallels that of normal neural stem cell cultures. Thus, the vast majority of SOX2− cells do not proliferate, as judged by Ki67 expression, and the expression of the gene dramatically decreases if the cells are kept under differentiating conditions, where they show signs of at least partial differentiation. We then hypothesized that SOX2 function may be conserved in glioblastoma TICs and therefore decided to study the effects of its silencing in glioblastoma primary cultures derived from seven patients, and chose to do so by knocking down SOX2 expression with a retroviral vector bearing an miRNA targeted to the coding region of the gene.
In principle, this approach should not be the best choice to target stem cells, which are thought to be rarely dividing cells. Given the requirement for cell division for retroviral infection, there is a risk of mainly targeting only proliferating precursors and not the true cancer stem cells, underscored by the observed strict correlation between the infection rate and proliferation rate of the different cultures. However, this was not the case, as mice injected with cells transduced with the control vector always developed many tumor masses positive for the reporter gene (GFP), showing that not only the fast proliferating more mature fraction, but also the true slower proliferating TICs, had been infected. This would indicate that, under these culture conditions, a sizable number of glioblastoma TICs divide at least once during the 4-day infection procedure. Thus, although the infection may be biased towards the faster proliferating precursors, the observed effect should be attributed to SOX2 silencing in both stem and precursor cells.
SOX2 knockdown in glioblastoma TICs did not have a dramatic effect in the short term. Thus, no induction of massive apoptosis, cell senescence, or differentiation could be detected in the SOX2 silenced cells. However, their numbers slowly but steadily decreased over time, leading to their virtual disappearance from the culture in about 5 weeks. Such an effect could be explained by a lengthening of the cell cycle that would dilute out the silenced population. However, all the data point to a gradual exit from the cell cycle of the SOX2 silenced cells. Indeed, the proportion of Ki67 cells decreased a few days after SOX2 knockdown. Accordingly, the size of the clones originating from these cells 1 week after infection was smaller than that of the corresponding controls 7 days after plating, and even smaller after 14 days, and when the cloning assay was performed with cells kept in culture for 4 weeks after infection, when the cultures still contained a residual GFP+ population, the silenced cells could not produce any clone at all.
Thus, some cells stopped proliferating right after SOX2 knockdown, whereas others engaged in a few rounds of cell division before becoming quiescent, and others still kept dividing longer, for up to 4 weeks after infection. In light of what has been previously observed in murine neural stem cells [9, 11], the most likely explanation is that, in the absence of SOX2, glioblastoma TICs loose a brake that prevents them from differentiating. The glioblastoma cells then become progressively more mature, exit from the cell cycle, and eventually die, disappearing from the culture. The proportion of cells maturing each day would be rather small, especially in the heterogeneous population of glioblastoma TICs, in which differentiation is impaired and several cells already express differentiation markers, explaining why we failed to clearly detect it. Although direct evidence of such an increase in the differentiation rate is difficult to obtain in TIC cultures, due to their impaired aberrant expression of differentiation markers, the expression of two transcription factors, Olig2 and BF1, typical of early neural precursor cells significantly decreased in SOX2 silenced TICs. Moreover, when SOX2 was silenced in normal mouse neural stem cells, an unusually high proportion of these cells not only exited from the cell cycle but also showed increased expression of a neuronal marker, indicating that they had become engaged in this differentiation pathway even under culture conditions that should prevent them from doing so.
SOX2 knockdown must have an effect not only on committed precursor cells, but also on the cancer stem cells contained in the TICs, as shown by the loss of tumorigenicity of the SOX2 silenced cells.
Similar results were obtained with a retroviral vector carrying a different miRNA, directed to the 3′-UTR of SOX2 mRNA, indicating that the observed effect was not due to off-target silencing of some other gene. Moreover, the silencing vector directed to the 3′-UTR of SOX2 mRNA has no effect on cells constitutively expressing SOX2 from a transgene lacking the 3′-UTR, which cannot be silenced by such a vector.
It then appears that SOX2 is also fundamental for the maintenance of the self-renewal capacity of neural stem cells when they have acquired cancer properties. This function must be essential because, in the absence of SOX2, the cells stop proliferating, notwithstanding the many mutations they have accumulated.
SOX2, or perhaps its immediate downstream effectors, would then be an ideal target for glioblastoma therapy.
Conclusion
We silenced SOX2 expression by RNA interference in TICs derived from glioblastoma patients. We demonstrated that SOX2 controls the proliferation of TICs as well as of normal murine neural stem cells. Silencing of SOX2 leads to loss of tumorigenicity of glioblastoma TICs, notwithstanding the many mutations they have accumulated. Our results demonstrate that glioblastoma TICs maintain stemness using the same mechanisms of normal neural stem cells. These observations support a hierarchical model of brain cancer controlled by SOX2 and open the way to find downstream genes as therapeutic targets.
Acknowledgments
We thank Daniele Reverberi and Fabrizio Loiacono, IST Oncologia Medica C, Genova, for help with the FACS sorting. Marzia Perera is the recipient of a FIRC fellowship.
This work was supported by grants from Ministero della Sanità, Associazione Italiana per la Ricerca sul Cancro (AIRC), Fondazione Carige, and MIUR-PRIN.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
Author notes
Author contributions: R.M.R.G.: conception, design, collection, analysis, and interpretation of data, manuscript writing; F.G.: collection and assembly of data, analysis and interpretation of data; D.M.: assembly, analysis, and interpretation of data; M.C.C.: collection of data; P.M.: provision of study material, analysis and interpretation of data; M.P.: collection and assembly of data; G.L.R.: provision of study materials; G.L.Z.: provision of study materials; A.D.: conception, analysis, and interpretation of data, manuscript writing; G.C.: conception, analysis, and interpretation of data, manuscript writing. A.D. and G.C. contributed equally to this work.
Disclosure of potential conflicts of interest is found at the end of this article.
First published online in STEM CELLS Express October 23, 2008.
Telephone: 39-010-5737404; Fax: 39-010-5737405
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}