1. Introduction
An expanding body of evidence demonstrates that chronic autoimmune inflammatory diseases are associated with accelerated atherosclerosis and increased cardiovascular morbidity and mortality compared to the general population [
1,
2]. Although rheumatoid arthritis has been most extensively studied, an abundance of data now exists demonstrating excess cardiovascular risk in a multitude of other inflammatory diseases, including systemic lupus erythematosus, the seronegative spondyloarthropathies, psoriasis and inflammatory bowel disease [
3,
4,
5,
6,
7,
8].
Endothelial dysfunction has been postulated to represent an initial step in the pathogenesis of atherosclerosis in the general population [
9]. Accordingly, efforts to elucidate unique mechanisms driving increased cardiovascular risk in patients with inflammatory diseases have often focused on the endothelium, which serves as an interface for multiple converging risk factors. In this review, we outline the evidence for and the significance of endothelial dysfunction in several chronic inflammatory diseases. We review the epidemiology and potential mechanisms of endothelial dysfunction in inflammatory diseases, highlighting shared features. Finally, we summarize the available data regarding the efficacy of anti-inflammatory therapies in reducing endothelial dysfunction and potentially mitigating cardiovascular risk.
We queried the PubMed database (NCBI, Bethesda, MD, USA) using the MESH searches for relevant studies using the following search terms in various combinations: rheumatoid arthritis; systemic lupus erythematosus; psoriasis; seronegative spondyloarthritis; inflammatory bowel disease; endothelial function; endothelial dysfunction; endothelial activation; forearm blood flow; flow-mediated vasodilation; cardiovascular disease (CVD); cardiovascular mortality; myocardial infarction; inflammation. Because of the limited number of relevant studies, there were no defined inclusion or exclusion criteria. Studies were screened informally for size and methodological quality. Studies reviewed ranged over the period of 1982–2014, with preference given to more recent data. Systematic reviews and meta-analyses were incorporated when available.
2. Endothelial Dysfunction: Definitions and Prognostic Implications
Through its capacity to sense and respond to mechanical and biochemical stimuli, the endothelium plays an active and critical role in the physiologic regulation of vascular tone, cellular adhesion, vascular smooth muscle migration and resistance to thrombosis [
9,
10]. Endothelial dysfunction—perhaps more appropriately referred to as endothelial activation—refers to its failure to perform these physiologic functions, often as a maladaptive response to pathological stimuli. The phenotypic features of endothelial dysfunction include upregulated expression of cellular adhesion molecules, compromised barrier function leading to increased leukocyte diapedesis, increased vascular smooth muscle tone secondary to impaired processing of vasodilator substances such as nitric oxide and prostacyclin as well as increased production of vasoconstrictor substances including endothelin, and reduced resistance to thrombosis [
10]. These processes are thought to represent important steps in the initiation and maintenance of atherosclerosis and have been associated with propensity towards atherothrombosis and cardiovascular complications in advanced disease [
9,
10,
11].
Endothelial dysfunction has emerged as an important surrogate endpoint for cardiovascular events. Its role in initiating the cascade of events leading to atherosclerosis and atherothrombosis may position it well for use as an early indicator of disease at a point that may allow for effective risk factor modification or pharmacologic intervention prior to the development of full-blown atherosclerosis. Furthermore, the endothelium is viewed as an integrator of vascular risk: the mechanisms by which epidemiologically proven cardiovascular risk factors lead to atherosclerosis might be interrogated best at the level of the endothelium, where the processing of these pathogenic signals may converge into one or several common pathways in the genesis of advanced atherosclerosis.
3. Assessment of Endothelial Function
Endothelial function can be assessed in humans by assaying its capacity to perform its various physiologic functions, including regulation of vasomotor tone, expression of adhesion molecules and maintenance of an anti-thrombotic microenvironment. In contemporary clinical research, endothelial function is typically assessed by measuring changes in vasomotor tone in response to various stimuli. Methods of measuring vascular reactivity have become the standard largely due to their reproducibility and demonstrated correlation with other measures of atherosclerosis. Quantification of soluble cellular adhesion molecule expression has also been widely performed, although the usefulness of this technique has not been well established. The most common methods are reviewed below.
3.1. Forearm Blood-Flow
Quantification of forearm blood-flow (FBF) by venous occlusion plethysmography in response to intra-arterial infusions of vasodilator substances has been historically used to assess vascular reactivity in various patient populations [
12]. In this method, endothelial-dependent vasodilation is assayed by intra-brachial infusion of acetylcholine (ACh), an endothelium-dependent vasodilator via induction of endothelial nitric oxide synthase (eNOS) and prostacyclin. The vasodilator response to sodium nitroprusside (SNP), a direct nitric oxide donor and endothelium-independent vasodilator, is also typically assessed in this method. Pure endothelial dysfunction is characterized by impaired vasodilation in response to ACh but intact responsiveness to SNP. FBF has been shown to correlate closely with coronary artery ACh-induced vasodilation [
13]. Although reproducible and accurate, FBF measurement is limited by its requirement for arterial cannulation, thereby limiting its repeatability and use in larger cohort studies.
3.3. Microvascular Vasodilation
There has been some concern that assessment of conduit artery function may not accurately reflect endothelial function in the microcirculation. Assessment of endothelium-dependent vasodilation in the cutaneous microcirculation is typically performed by using laser Doppler imaging to measure responses to infusion of vasodilator substances via iontophoresis [
18]. Similar to assessment of the larger vessels, ACh is used as the endothelium-dependent vasodilator while SNP is used to assess endothelium-independent mechanisms. These substances are delivered transdermally by application of an electrical field to induce migration of the ionized drug into cutaneous capillaries. Laser Doppler imaging allows for measurement of microvascular perfusion. Various other techniques have been employed to assess microvascular function in tissues other than the skin. Transthoracic echocardiography has been used to assess coronary flow reserve and recently positron emission tomography (PET) has been used to assess myocardial blood flow and coronary flow reserve [
19]. These techniques have not yet been widely applied to assessment of endothelial function in patients with chronic inflammatory diseases.
3.4. Plasma Biomarkers of Endothelial Dysfunction
Efforts to define plasma biomarkers for endothelial dysfunction have largely focused on soluble intercellular adhesion molecules (CAMs), including intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule (VCAM-1), E-selectin and others [
20,
21]. These molecules are typically expressed at the surface of the endothelial cell in response to activation by inflammatory cytokines or other stimuli and bind leukocyte-specific adhesion molecules, leading to increased leukocyte affinity to the endothelial surface and eventually increased transendothelial migration. Although they have been extensively studied, the prognostic value of soluble CAMs remains limited due to poor reproducibility. There is some evidence, however, that elevated ICAM-1 and E-selectin levels are associated with increased risk of incident clinical coronary heart disease [
21].
Recently, the protein asymmetric dimethylarginine (ADMA), an endogenous eNOS inhibitor, has garnered interest as a potential biomarker for endothelial dysfunction [
22]. Plasma levels of ADMA are negatively correlated with NO levels and are elevated in a variety of diseases traditionally associated with cardiovascular risk, including hypertension, dyslipidemia, diabetes mellitus and chronic kidney disease [
22]. Elevated plasma ADMA has also been associated with increased risk of cardiovascular events across a range of patient populations [
23]. Numerous other molecules, including inflammatory cytokines, regulators of thrombosis and indicators of endothelial damage and repair have been proposed as biomarkers for endothelial dysfunction [
24]. The clinical significance of most of these potential biomarkers remains unclear, however.
4. Clinical Findings of Major Chronic Inflammatory Diseases Suggest Cardiovascular Risk
Rheumatoid arthritis, systemic lupus erythematosus, the seronegative spondyloarthropathies, psoriasis and inflammatory bowel disease have all been associated clinically with excessive cardiovascular risk [
1,
2,
3,
4,
5,
6,
7,
8,
25]. Over the last several decades, there has been considerable interest in characterizing this excess cardiovascular risk in an attempt to identify potential risk factors and mechanisms responsible for the genesis of atherosclerosis in these populations (
Table 1).
Table 1.
Relative risk of cardiovascular morbidity and mortality.
Table 1.
Relative risk of cardiovascular morbidity and mortality.
| Disease | CAD Risk (RR or OR) | Cardiovascular Mortality (RR) |
|---|
| Rheumatoid Arthritis | 1.5–2.0 [26,27] | 1.5 [28] |
| Systemic Lupus Erythematosus | 2.2–2.6 [29,30] | 1.7 [31] |
| Psoriasis (severe) | 1.5–7.1 [7,25] | 1.1–1.6 [7,25] |
| Ankylosing Spondylitis | 1.9 [32,33] | 1.3–2.1 [5] |
| Inflammatory Bowel Disease | 1.2–1.4 [6,34] | 1.0 [34,35] |
4.1. Rheumatoid Arthritis (RA)
It has been known for many years that coronary artery disease is largely responsible for the excess morbidity and mortality in patients with RA. Endothelial dysfunction in RA was first described in a seminal 2002 study demonstrating impaired brachial artery responsiveness to ACh by FBF in patients with early disease [
36]. Impaired endothelium-dependent vasodilation has since been repeatedly demonstrated at various stages of disease and across a spectrum of disease activity by several different techniques [
37,
38,
39,
40,
41]. Microvascular dysfunction has similarly been described in RA, and endothelial-dependent vasodilation in the cutaneous microcirculation has been shown to correlate with disease activity [
42,
43]. There has been less consistency in the correlation between disease activity and macrovascular function, however. Whereas several studies have demonstrated impaired FMD or FBF in patients with early RA with low disease activity [
36,
37,
39,
40], others have failed to show differences in this population [
44,
45]. Discordance may be related to definitions of “early” and “low” disease activity. A 2012 systematic review of vascular function and morphology in RA included 57 cross-sectional studies and 27 longitudinal studies [
46]. The vast majority of these studies reported that endothelium-dependent vasodilation was significantly impaired in patients with RA compared to healthy controls. Studies addressing the correlation between endothelial function and markers of systemic inflammation and disease activity (tender/swollen joint counts, biomarkers of systemic inflammation) were less convincing, however. The authors concluded that the available evidence does not wholly support a correlation between disease activity and macrovascular function [
46].
Efforts to characterize endothelial function by measuring soluble plasma biomarkers in patients with rheumatoid arthritis have been largely unsuccessful. Littler
et al. [
47] first described an expression profile of intercellular adhesion molecules in 22 patients with RA. While ICAM-1, ICAM-3, VCAM-1, L-selectin and P-selectin were found to be elevated in sera of patients with RA, only P-selectin correlated with disease activity. Others have identified unique expression profiles in RA patients [
48,
49,
50], although ICAM-1 and P-selectin were also found to be elevated in RA patients in these studies. Several investigators have failed to demonstrate differences in adhesion molecule expression between patients and healthy controls [
51]. There is also discordance with regard to the correlation between adhesion molecule expression and markers of disease activity. Plasma levels of ADMA have also been found to be elevated in patients with RA. ADMA levels correlate inversely with FMD and directly with markers of systemic inflammation [
52]. In general, the clinical utility of biomarkers for endothelial dysfunction in inflammatory diseases remains unclear. While it appears unlikely that cellular adhesion molecules will serve as important prognostic indicators for CVD, ADMA is more promising. Other biomarkers currently under investigation, such as circulating endothelial progenitor cells, may prove to be useful markers of endothelial dysfunction.
4.2. Systemic Lupus Erythematosus (SLE)
The excess burden of CVD in patients with SLE is now well established. Similar to RA, endothelial function has been widely used as a surrogate endpoint for CVD in patients with SLE. Impaired FMD was observed in patients with SLE as early as 2002 [
53]. Multiple subsequent studies have validated this observation [
54,
55,
56], including studies interrogating endothelial function in the microcirculation [
57]. One study failed to demonstrate differences in FMD between SLE patients and controls, however [
58]. Differences in population characteristics may account for this discordance. Importantly, all of these studies excluded patients with known CVD. Taken together, the available evidence strongly supports the presence of impaired endothelium-dependent vasodilation in patients with SLE without documented CVD.
As with RA, efforts to characterize the expression profile of biomarkers for endothelial dysfunction in patients with SLE have been less successful than vascular reactivity studies. Sfikakis demonstrated increased levels of circulating ICAM-1 in patients with SLE [
59]. Tulek and colleagues replicated these results but failed to demonstrate a correlation between ICAM-1 levels and disease activity or markers of systemic inflammation [
60]. In contrast, Machold and colleagues failed to demonstrate differences in ICAM-1 levels between SLE patients and healthy controls [
61]. Several other groups have attempted to correlate adhesion molecule levels with markers of disease activity. The results have been widely variable, although at least two studies demonstrated a correlation between VCAM-1 levels and disease activity [
62,
63,
64]. Given the heterogeneity between studies and the disparate patterns of results, it is difficult to conclude that patients with SLE exhibit a distinct profile of adhesion molecule expression. There is some weak evidence, however, that during periods of high disease activity and increased systemic inflammation, levels of soluble intercellular adhesion molecules tend to be elevated in patients with SLE. The implications of these findings remain unclear.
4.3. The Seronegative Spondyloarthropathies and Psoriasis
Much less is known about the cardiovascular risk associated with the seronegative spondyloarthropathies; however, the available evidence suggests that these diseases confer increased risk of cardiovascular morbidity and mortality [
4,
5]. Similarly, only a handful of authors have assessed endothelial function in these diseases. Findings have been consistent, however. In two studies of patients with ankylosing spondylitis and one study with psoriatic arthritis, FMD was significantly impaired compared to healthy controls [
65,
66,
67]. One study demonstrated that the reduction in FMD correlated significantly with markers of disease activity [
65].
Less is known about adhesion molecule expression in this population. As with RA and SLE, the available data are discordant. Wendling
et al. [
68] demonstrated that mildly elevated ICAM-1 levels correlated with markers of disease activity (
i.e., erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), disease severity score) in patients with spondyloarthropathies. Moreover, markedly elevated ICAM-1 levels (>400) were found only in patients with disease. In contrast, Sari and colleagues failed to demonstrate any significant differences in ICAM, VCAM, E-selectin or P-selectin levels in patients with spondyloarthropathies [
69]. Plasma levels of ADMA also appear to be elevated in patients with spondyloarthropathies, although attempts to correlate ADMA levels with markers of disease activity or other markers of endothelial dysfunction have been unsuccessful [
70,
71].
Cutaneous psoriasis, even in the absence of joint disease, has been linked to accelerated atherosclerosis. The evidence for this association is less robust than for RA and SLE, though the available data support a significantly increased risk for cardiovascular events and mortality in patients with severe disease. Patients with mild disease may be spared [
7,
25]. Accordingly, many groups have sought to characterize endothelial function in patients with cutaneous psoriasis. Although results have been mixed, a 2014 systematic review of 20 studies of endothelial function in psoriasis patients found that FMD was significantly impaired in the majority of studies in which FMD was used [
72]. Some data suggest that the likelihood of endothelial dysfunction is correlated with disease severity or disease duration [
73]; however, a number of studies of patients with disease classified as “severe” have demonstrated no impairment in FMD [
74,
75]. Interestingly, increased carotid intima-media thickness, a measure of subclinical atherosclerosis, has been demonstrated repeatedly in patients with psoriasis and has been shown to correlate with FMD [
72,
73]. Taken together, these data suggest that patients with psoriasis display impaired endothelial-dependent relaxation and that this may correlate with future development of atherosclerosis and cardiovascular events.
4.4. Inflammatory Bowel Disease (IBD)
Similar to psoriasis, there has been recent interest in exploring the cardiovascular implications of inflammatory bowel diseases (IBD), Crohn’s disease and ulcerative colitis (UC). While individual studies have demonstrated an increased risk of cardiovascular events in patients with IBD, mortality risk is less clear. A 2014 meta-analysis of 9 studies demonstrated that patients with IBD were at significantly increased risk of ischemic heart disease and stroke [
6]. Mortality was not addressed. Multiple other groups, including Dorn and colleagues in a 2007 meta-analysis of 11 studies, have failed to demonstrate increased risk of cardiovascular mortality in patients with IBD [
35]. While there is ample evidence of accelerated atherosclerosis in this population, it remains unclear why these patients are spared from an associated mortality risk.
In 2003 Hatoum and colleagues demonstrated that the intestinal microvessels in patients with IBD show significantly impaired endothelium-dependent vasodilation compared to vessels from healthy control patients [
76]. In addition, the microcirculation in uninvolved segments of bowel remained unaffected, indicating that the mechanism of local endothelial dysfunction may not be related to systemic inflammation. This led to an exploration of generalized endothelial function in patients with IBD. Kocaman and colleagues first demonstrated that generalized endothelial dysfunction is a feature of ulcerative colitis [
77]. FMD of the brachial artery was significantly impaired in ulcerative colitis patients, and disease severity was an important determinant of the degree of impairment. Multiple subsequent studies have supported the observation that FMD of the brachial artery is impaired in patients with both UC and IBD, lending credence to the hypothesis that generalized endothelial dysfunction is a feature of IBD, not a tissue-specific phenomenon limited to the GI tract [
78,
79].
5. Animal Models
Multiple models of inflammatory arthritis have been described for the study of rheumatoid arthritis and other inflammatory joint diseases in animals. The collagen-induced arthritis (CIA) model in mice is the most widely-used animal model for the study of RA. In this model, intradermal inoculation of mice with type II collagen and Complete Freund’s Adjuvant (CFA) generates an intense inflammatory polyarthritis immunologically similar to RA [
80]. He and colleagues recently demonstrated that CIA mice have significantly impaired endothelium-dependent vasodilation compared to control mice [
81]. In a separate study, aortic endothelium isolated from CIA mice was shown to over-express VCAM-1 [
82]. Rats with a similar adjuvant-induced arthritis also exhibit both an inflammatory polyarthritis and impaired endothelium-dependent vasodilation [
83,
84]. A collagen-induced arthritis model was recently described in sheep. Sheep with inflammatory arthritis demonstrated marked impairment in endothelium-dependent vasodilation in the coronary and digital arteries, whereas endothelium-independent vasodilation was intact [
85]. Taken together, there is convincing evidence that animal models of inflammatory arthritis exhibit generalized endothelial dysfunction that may provide insight into the mechanisms linking inflammatory diseases and endothelial dysfunction in humans.
6. Mechanisms of Endothelial Dysfunction
While it is clear that multiple chronic inflammatory diseases are associated with endothelial dysfunction and cardiovascular morbidity, the mechanistic links between inflammatory diseases and CVD have not been fully elucidated. The role of traditional cardiovascular risk factors in patients with RA and SLE has received considerable attention, though traditional factors alone are insufficient to explain the excess burden of CVD in these populations. It seems likely that chronic inflammation, a shared feature of these diseases, is involved in the pathogenesis of accelerated endothelial dysfunction. Several potential mechanisms are explored below (
Figure 1).
Figure 1.
Mediators of endothelial dysfunction in inflammatory diseases. TNF-α (tumor necrosis factor-α) exerts its effects on the endothelium through its receptor, TNFR. Binding of TNFR by TNF-α leads to diminished eNOS (endothelial nitric oxide synthase) protein expression via suppression of promoter activity and destabilization of its mRNA. TNFR suppresses eNOS activity by preventing the degradation of its endogenous inhibitor, ADMA (asymmetric dimethylarginine). TNFR signaling also induces the transcription factor NF-κB leading to enhanced expression of intercellular adhesion molecules (ICAM-1: intercellular adhesion molecule-1; VCAM-1: vascular cell adhesion molecule-1), TNF-α and Nox1 (NADPH-oxidase-1). NF-κB induction is also mediated by oxidized low density lipoprotein (oxLDL), reactive oxygen species (ROS) and binding of various autoantibodies (AECA: anti-endothelial cell antibodies; APLA: antiphospholipid antibodies; anti-oxLDL: anti-oxidized LDL antibodies). eNOS uncoupling, mediated in part by ROS, is associated with reduced NO (nitric oxide) production and enhanced generation of ROS. eNOS activity is also suppressed by oxLDL.
Figure 1.
Mediators of endothelial dysfunction in inflammatory diseases. TNF-α (tumor necrosis factor-α) exerts its effects on the endothelium through its receptor, TNFR. Binding of TNFR by TNF-α leads to diminished eNOS (endothelial nitric oxide synthase) protein expression via suppression of promoter activity and destabilization of its mRNA. TNFR suppresses eNOS activity by preventing the degradation of its endogenous inhibitor, ADMA (asymmetric dimethylarginine). TNFR signaling also induces the transcription factor NF-κB leading to enhanced expression of intercellular adhesion molecules (ICAM-1: intercellular adhesion molecule-1; VCAM-1: vascular cell adhesion molecule-1), TNF-α and Nox1 (NADPH-oxidase-1). NF-κB induction is also mediated by oxidized low density lipoprotein (oxLDL), reactive oxygen species (ROS) and binding of various autoantibodies (AECA: anti-endothelial cell antibodies; APLA: antiphospholipid antibodies; anti-oxLDL: anti-oxidized LDL antibodies). eNOS uncoupling, mediated in part by ROS, is associated with reduced NO (nitric oxide) production and enhanced generation of ROS. eNOS activity is also suppressed by oxLDL.
![Ijms 15 11324 g001]()
6.1. Tumor Necrosis Factor-α (TNF-α)
The vascular endothelium is known to be a target of TNF-α. On a cellular level, TNF-α induces the expression of genes associated with inflammation, coagulation and proliferation. Decreased nitric oxide (NO) bioavailability appears to be a common and critical step linking TNF-α to endothelial dysfunction. Multiple groups have shown that eNOS protein expression is reduced via TNF-α- induced inhibition of eNOS promoter activity and mRNA destabilization [
86,
87]. NO availability is also compromised in the presence of TNF-α secondary to impaired degradation of ADMA, an endogenous inhibitor of NOS. Furthermore, TNF-α induces CAM expression on the surface of vascular endothelial cells. This effect is mediated via isoform one of the TNF-receptor (TNFR1). Activation of TNFR1 leads to increased CAM expression via induction of NF-κB [
87,
88]. NO is also known to be an inhibitor of CAM expression [
89]. TNF-α may therefore lead to increased CAM expression by multiple pathways.
The effect of TNF-α on NO availability and subsequent endothelial dysfunction has also been demonstrated
in vivo in both animal and human models. Intravenous delivery of TNF-α in rats leads to impaired endothelium-dependent vasodilation [
90]. Intra-arterial infusion of TNF-α in humans also impairs local endothelium-dependent vasodilation measured by FBF [
91]. Non-specific induction of an acute systemic inflammatory response by
Salmonella typhi vaccination also causes reduced FBF [
92]. This effect is mediated by impaired NO bioavailability as demonstrated by rescue of vascular reactivity with the NOS inhibitor
l-NNMA (
l-N
G-monomethyl Arginine) [
91]. The downstream effects of TNF-α-mediated inflammation are illustrated in an
apoE−/−, TNF-α−/− mouse model. Mice deficient in TNF-α develop less atherosclerosis than those with intact TNF-α expression (
i.e.,
apoE−/− single knockout) [
93]. This is associated with decreased expression of ICAM-1, VCAM-1 and monocyte chemotactic protein-1 (MCP-1).
It is well known that TNF-α plays a critical role in the inflammation associated with RA, SLE, IBD, psoriasis and spondyloarthritis. This common feature is illustrated by the efficacy of anti-TNF-α agents in many of these diseases. Given the central role of TNF-α in the pathogenesis of many chronic inflammatory diseases and its well-characterized effects on the endothelium as described above, it is reasonable to conclude that increased circulating TNF-α is implicated in the induction of endothelial dysfunction and initiation of atherosclerosis in these diseases (
Figure 2). This hypothesis is supported by the beneficial effects of anti-TNF-α agents on endothelial function in patients with chronic inflammatory diseases, as discussed later.
6.2. Oxidative Stress
Chronic inflammatory diseases are generally associated with increased oxidative stress. In RA, reactive oxygen species (ROS) levels from peripheral blood neutrophils correlate positively with disease severity and markers of systemic inflammation [
94]. Inflammatory cytokines, including TNF-α, are largely responsible for the increased ROS production in these diseases. TNF-α increases activity of the NADPH oxidases (NOX), which catalyze the transfer of electrons onto molecular oxygen to generate superoxide by neutrophils and endothelial cells [
87,
95]. As discussed previously, the bioavailability of NO is a critical factor in determining vascular reactivity. In addition to its production by NOS and metabolism by ADMA, NO bioavailability is also modulated by ROS. Superoxide rapidly reacts with NO to produce peroxynitrite, thereby decreasing NO availability [
96]. The importance of this mechanism is demonstrated by observations that eNOS is paradoxically up-regulated in hypertension and diabetes mellitus, conditions associated with endothelial dysfunction [
96]. ROS also contribute to the “uncoupling” of eNOS, leading to enhanced superoxide generation and decreased NO production [
97]. Multiple
in vivo animal models have demonstrated reduced NO bioavailability in the presence of elevated ROS, and reversal of endothelial dysfunction has been achieved via infusion of antioxidants [
98].
In addition to downregulating NO bioavailability, superoxide and other ROS are capable of inducing NF-κB, a critical step in transforming endothelial cells into an “activated” state characterized in part by increased surface expression of CAMs [
94,
99]. As discussed previously, CAM expression by endothelial cells represents a fundamental feature of endothelial dysfunction, leading to enhanced leukocyte affinity and eventually migration into the subendothelial space, key steps in the initiation and maintenance of atherosclerosis. Activation of NF-κB can also stimulate NOX expression, further enhancing ROS production in the endothelium and regenerating the destructive loop of inflammation and oxidative stress [
100].
Figure 2.
From local inflammation to systemic endothelial dysfunction. TNF and inflammatory cytokines spread from the primary, disease-specific site of local inflammation into the systemic circulation to propagate a systemic inflammatory response. The byproducts of systemic inflammation, including reactive oxygen species (ROS), lipid abnormalities and other metabolic derangements are dependent on peripheral tissues such as the liver and adipose. These mediators elicit independent and complementary effects on the endothelium, leading to a state of endothelial dysfunction characterized by increased adhesion molecule expression (VCAM, ICAM), leukocyte diapedesis, ROS production and decreased NO (nitric oxide)-mediated smooth muscle relaxation and vascular dilation. Autoantibodies are generated in a disease-specific manner and induce similar changes in endothelial function.
Figure 2.
From local inflammation to systemic endothelial dysfunction. TNF and inflammatory cytokines spread from the primary, disease-specific site of local inflammation into the systemic circulation to propagate a systemic inflammatory response. The byproducts of systemic inflammation, including reactive oxygen species (ROS), lipid abnormalities and other metabolic derangements are dependent on peripheral tissues such as the liver and adipose. These mediators elicit independent and complementary effects on the endothelium, leading to a state of endothelial dysfunction characterized by increased adhesion molecule expression (VCAM, ICAM), leukocyte diapedesis, ROS production and decreased NO (nitric oxide)-mediated smooth muscle relaxation and vascular dilation. Autoantibodies are generated in a disease-specific manner and induce similar changes in endothelial function.
6.3. Dyslipidemia
The role of traditional cardiovascular risk factors such as dyslipidemia and insulin resistance in the pathogenesis of endothelial dysfunction and atherosclerosis in patients with chronic inflammatory diseases has received significant attention. Although it has been reported that patients with RA and other rheumatic diseases are more likely to have elevated low-density lipoprotein (LDL) and total cholesterol and reduced high-density lipoprotein (HDL) levels, the data are inconsistent. For example, multiple studies have shown no differences in lipid profiles of patients with RA
versus healthy controls, whereas others have described a distinct profile of suppressed LDL and HDL in RA patients with more advanced disease (
i.e., rheumatoid cachexia) [
101]. Chronic inflammation structurally alters lipoproteins in ways that are not reflected in standard lipid profiles, however. Inflammation has been shown to modify LDL into small, dense particles that are known to be pro-atherogenic. Indeed, RA patients have elevated plasma levels of small, dense LDL particles [
102]. TNF-α also enhances the oxidative modification of LDL by increasing ROS production. In addition, HDL is modified by inflammation. Small HDL particles, known to play a critical role in reverse-cholesterol transport, have been shown to be decreased in patients with RA. The mechanisms by which small HDL is regulated have been extensively reviewed elsewhere [
101].
Dyslipidemia is independently associated with endothelial dysfunction. Elevated LDL and total cholesterol are associated with impaired endothelium-dependent vasodilation, whereas elevated HDL levels correlate with improved endothelial function [
103]. Impaired endothelial function in dyslipidemic patients may be caused by reduced NO availability. In dyslipidemic patients, NO availability may be impaired by oxidized LDL-mediated reduction in NOS activity or by enhanced metabolism of NO by ADMA [
104]. Lipoproteins are also implicated in ROS production via modulation of NOX activity and by contributing to the “uncoupling” of eNOS [
105]. In addition to modulation of NO and ROS production, oxidized LDL induces upregulation of CAM expression at the endothelial surface and secretion of TNF-α via induction of NF-kB. These mechanisms are reviewed elsewhere, and additional mechanisms of LDL-mediated endothelial dysfunction have been described in various models [
105].
6.4. Autoantibodies
Many chronic inflammatory diseases are associated with production of autoantibodies, many of which are instrumental in the pathogenesis of the disease. Similarly, autoantibodies directed against normal endothelial or plasma constituents have been detected and implicated in the pathogenesis of endothelial dysfunction and atherosclerosis in the general population. Anti-endothelial cell antibodies (AECA) directed against a variety of endothelial cell structural proteins have been identified in a number of autoimmune diseases, including SLE [
106]. These antibodies have been implicated in the pathogenesis of lupus-associated vasculitis and induce endothelial dysfunction via induction of NF-kB, leading to upregulation of CAMs and inflammatory cytokines [
107]. Although these antibodies have been described in SLE and vasculitis, their roles, if any, in the genesis of systemic endothelial dysfunction in SLE and other inflammatory diseases, remain unclear.
Antibodies directed against oxidized LDL (anti-oxLDL) have been described in patients with and without chronic inflammatory diseases. In SLE, anti-oxLDL antibodies correlate with disease activity and markers of systemic inflammation [
108]. Although anti-oxLDL antibodies have been correlated with markers of atherosclerosis in various models, their impact on endothelial cell function remains to be elucidated. There is some evidence that anti-phospholipid antibodies may exhibit cross-reactivity with oxLDL [
106]. This would provide a viable mechanism for induction of endothelial dysfunction in patients with SLE and antiphospholipid antibodies.
Antiphospholipid antibodies (aPLs) are present in the serum of nearly one third of lupus patients. The antiphospholipid antibody syndrome (APS) is characterized by recurrent venous or arterial thrombosis, pregnancy loss and the presence of antiphospholipid antibodies (
i.e., lupus anticoagulant, anticardiolipin antibody, and anti-B2GPI antibody). Although their contribution to venous and arterial thrombotic events is well known, the relative contribution of aPL to the development of endothelial dysfunction in humans, if any, is currently unclear. The effect of aPL on endothelium-dependent vasodilatation may be reflected in the observation that patients with aPL exhibit impaired FMD and reduced NO bioavailability
versus healthy controls [
109]. aPL have also been shown to enhance CAM expression at the endothelial surface
in vitro and
in vivo. Efforts to measure circulating levels of soluble adhesion molecules in patient with APS have been less consistent, however [
106]. Further studies are required to clarify whether aPL are responsible for inducing endothelial dysfunction and atherosclerosis in the absence of other complicating risk factors.
7. Effects of Pharmacologic Interventions on Endothelial Function
7.1. Anti-Inflammatory Therapy
Methotrexate remains the mainstay of therapy for RA and several other rheumatic diseases (
Table 2). An inhibitor of folic acid metabolism, methotrexate sharply reduces systemic inflammation and dramatically improves synovitis in patients with inflammatory arthritis. Methotrexate also appears to improve endothelium-dependent vasodilation in patients with RA, although the data are limited [
2,
110]. Inhibitors of TNF-α have been employed with increasing frequency for patients with a variety of inflammatory diseases, including RA, spondyloarthritis, IBD and psoriasis.
The critical role of TNF-α in the generation of severe systemic inflammation in these conditions likely explains the effectiveness of these agents. TNF-α may also be largely responsible for the endothelial dysfunction and accelerated atherosclerosis in these patients, making anti-TNF-α agents attractive therapeutic options for preventing CVD in this population. Numerous studies have demonstrated improved endothelium-dependent vasodilation in patients with RA after initiation of anti-TNF-α therapy. This has been demonstrated in a vessel-specific manner by measuring FBF immediately after intra-brachial infusion of infliximab. In this model Cardillo and colleagues demonstrated that the local effect of infliximab on the brachial artery improved brachial artery endothelial function without altering markers of systemic inflammation [
111]. Multiple other studies have demonstrated that anti-TNF-α agents improve FMD in RA patients who are refractory to conventional disease modifying anti-rheumatic drugs (DMARD) therapy [
112,
113,
114,
115,
116,
117]. Anti-TNF-α agents also improve endothelium-dependent vasodilation in patients with spondyloarthritis [
118,
119], cutaneous psoriasis [
72,
120] and IBD [
121], although studies are small and few. Improvement in endothelial function with anti-TNF-α therapy may correlate with improvement in disease activity and markers of systemic inflammation [
122]. The duration of the response has been controversial, however. Several studies in RA have shown that anti-TNF-α agents induce a rapid improvement in FMD that is lost after a period of weeks despite effective control of disease activity and systemic inflammation [
114,
116]. Other studies have demonstrated sustained improvements in endothelial function [
115,
123]. Factors contributing to differences in duration of response remain unclear.
Table 2.
Medication and Effect on Endothelial Function.
Table 2.
Medication and Effect on Endothelial Function.
| Medication | Disease(s) | Target | Effect on Endothelial Function |
|---|
| Methotrexate | RA, SpA, Psoriasis | Folic acid metabolism, lymphocyte proliferation, inflammation | Likely beneficial [2,110] |
| Anti-TNF-α agents | RA, SpA, Psoriasis, IBD | TNF-α-mediated inflammation | Strong evidence for benefit [72,112,113,114,115,116,117,118,119,121] |
| Corticosteroids | RA, SpA, IBD, SLE | Spectrum of immune and inflammatory responses | Inconclusive [124,125] |
| Statins | RA, SLE, traditional CVD risk factors | LDL, eNOS, pleiotropic effects on inflammation | Strong evidence for benefit [126,127,128,129] |
Anti-TNF-α agents have also been shown to reduce levels of plasma biomarkers of endothelial dysfunction, although results have been inconsistent. Klimiuk
et al. [
130] demonstrated that etanercept administration reduced levels of soluble ICAM-1, VCAM-1 and E-selectin in patients with RA. Gonzalez-Gay and colleagues found reductions only in soluble ICAM-3 and P-selectin after infliximab infusions for patients with RA [
131]. Adalimumab therapy in patients with psoriasis has been shown to reduce ICAM-1 levels without affecting other CAMs [
120]. These findings are similar to results from studies examining levels of CAMs at baseline across various inflammatory diseases: it has been difficult to discern a consistent profile of CAM expression prior to or in response to disease-modifying therapy. Although CAM expression may be a general marker for systemic inflammation and endothelial dysfunction, its utility in clinical and translational research may be limited.
Corticosteroids have long been used to manage a variety of inflammatory diseases, but their effects on CVD have been controversial. The association between steroids and insulin resistance and obesity has raised concern for increased cardiovascular risk, while their anti-inflammatory effects might mitigate this risk. Studies addressing the association between long-term steroid use in RA and CVD have yielded variable results. A 2011 systematic review of studies of low-dose steroid use in RA found that corticosteroids are generally associated with mildly increased cardiovascular risk [
132]. Studies did not reveal an effect of steroids on markers of subclinical atherosclerosis and endothelial function, however. Other observational studies have demonstrated an association between corticosteroid use and lower rates of subclinical atherosclerosis compared to patients not using steroids [
133]. Veselinovic and colleagues demonstrated that FMD is higher in RA patients treated with corticosteroids
versus no therapy [
124]. This study conflicts with a randomized prospective study, by Hafstrom, showing that addition of steroids to DMARD therapy does not improve endothelial function in RA patients [
125]. These results highlight the difficulty of studying the effects of single-agent steroid therapy on patients with inflammatory disease in the modern era. Measuring the added benefit of steroids in the context of background immune-suppressing therapy is unlikely to reveal significant improvements, even if corticosteroids may have this effect in isolation.
8. Conclusions
Patients with chronic inflammatory diseases are at high risk for cardiovascular morbidity and mortality. In many inflammatory diseases, this heightened risk of CVD is reflected in early endothelial dysfunction as assessed by vasoreactivity studies, even in the absence of detectable atherosclerosis. The endothelium therefore represents an integrator of vascular risk and the study of its dysfunction may help elucidate mechanisms driving accelerated atherosclerosis in these populations.
There is strong evidence that the mechanisms responsible for accelerated atherosclerosis in patients with inflammatory diseases are related to the high-grade inflammation inherent to the primary disease process. The effects of TNF-α and inflammatory cytokines on induction of endothelial dysfunction are well described and are likely to represent key mediators of endothelial dysfunction and atherosclerosis. Furthermore, the numerous studies demonstrating improved endothelial function after anti-TNF-α therapy highlight the importance of these molecules in the pathogenesis of endothelial dysfunction and may lead the way toward advances in pharmacologic prevention of CVD in these populations. Many other mechanisms, including autoantibodies, oxidative stress and interactions with traditional risk factors such as dyslipidemia and insulin resistance are likely to be involved, and further research is required to elucidate the relative importance of these processes. Finally, current strategies to reduce cardiovascular morbidity and mortality are focused on controlling traditional modifiable cardiovascular risk factors and reducing disease activity. The precise mechanisms driving atherosclerosis are likely to vary between different inflammatory diseases, however, and parsing out these subtleties may help identify unique therapeutic targets for each disease.
Acknowledgments
This work was supported by the Office of Research and Development, Department of Veterans Affairs 1BX001729 to F.J.M. and the American Heart Association GRNT12060205 to F.J.M. The funding sources had no role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spano, F.; Puppo, F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012, 224, 309–317. [Google Scholar] [CrossRef]
- Prati, C.; Demougeot, C.; Guillot, X.; Godfrin-Valnet, M.; Wendling, D. Endothelial dysfunction in joint disease. Jt. Bone Spine 2014. [Google Scholar] [CrossRef]
- Nikpour, M.; Gladman, D.D.; Urowitz, M.B. Premature coronary heart disease in systemic lupus erythematosus: What risk factors do we understand? Lupus 2013, 22, 1243–1250. [Google Scholar] [CrossRef]
- Peters, M.J.; van der Horst-Bruinsma, I.E.; Dijkmans, B.A.; Nurmohamed, M.T. Cardiovascular risk profile of patients with spondylarthropathies, particularly ankylosing spondylitis and psoriatic arthritis. Semin. Arthritis Rheum. 2004, 34, 585–592. [Google Scholar] [CrossRef]
- Mathieu, S.; Motreff, P.; Soubrier, M. Spondyloarthropathies: an independent cardiovascular risk factor? Jt. Bone Spine 2010, 77, 542–545. [Google Scholar] [CrossRef]
- Singh, S.; Singh, H.; Loftus, E.V., Jr.; Pardi, D.S. Risk of cerebrovascular accidents and ischemic heart disease in patients with inflammatory bowel disease: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2014, 12, 382–393 e381. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Neimann, A.L.; Shin, D.B.; Wang, X.; Margolis, D.J.; Troxel, A.B. Risk of myocardial infarction in patients with psoriasis. JAMA 2006, 296, 1735–1741. [Google Scholar] [CrossRef]
- Rosner, S.; Ginzler, E.M.; Diamond, H.S.; Weiner, M.; Schlesinger, M.; Fries, J.F.; Wasner, C.; Medsger, T.A., Jr.; Ziegler, G.; Klippel, J.H.; et al. A multicenter study of outcome in systemic lupus erythematosus. II. Causes of death. Arthritis Rheum. 1982, 25, 612–617. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef]
- Bonetti, P.O. Endothelial Dysfunction: A Marker of Atherosclerotic Risk. Arterioscler Thromb Vasc Biol 2002, 23, 168–175. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Lind, L.; Hall, J.; Larsson, A.; Annuk, M.; Fellstrom, B.; Lithell, H. Evaluation of endothelium-dependent vasodilation in the human peripheral circulation. Clin. Physiol. 2000, 20, 440–448. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar]
- Ghiadoni, L.; Salvetti, M.; Muiesan, M.L.; Taddei, S. Evaluation of endothelial function by flow mediated dilation: Methodological issues and clinical importance. High Blood Press Cardiovasc. Prev. 2014, in press. [Google Scholar]
- Anderson, T.J.; Uehata, A.; Gerhard, M.D.; Meredith, I.T.; Knab, S.; Delagrange, D.; Lieberman, E.H.; Ganz, P.; Creager, M.A.; Yeung, A.C.; et al. Close relation of endothelial function in the human coronary and peripheral circulations. J. Am. Coll. Cardiol. 1995, 26, 1235–1241. [Google Scholar] [CrossRef]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef]
- Inaba, Y.; Chen, J.A.; Bergmann, S.R. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: A meta-analysis. Int. J. Cardiovasc. Imaging 2010, 26, 631–640. [Google Scholar] [CrossRef]
- Roustit, M.; Cracowski, J.L. Non-invasive assessment of skin microvascular function in humans: An insight into methods. Microcirculation 2012, 19, 47–64. [Google Scholar] [CrossRef]
- Recio-Mayoral, A.; Mason, J.C.; Kaski, J.C.; Rubens, M.B.; Harari, O.A.; Camici, P.G. Chronic inflammation and coronary microvascular dysfunction in patients without risk factors for coronary artery disease. Eur. Heart J. 2009, 30, 1837–1843. [Google Scholar] [CrossRef]
- Ridker, P.M.; Hennekens, C.H.; Roitman-Johnson, B.; Stampfer, M.J.; Allen, J. Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men. Lancet 1998, 351, 88–92. [Google Scholar]
- Hwang, S.J.; Ballantyne, C.M.; Sharrett, A.R.; Smith, L.C.; Davis, C.E.; Gotto, A.M., Jr.; Boerwinkle, E. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the Atherosclerosis Risk In Communities (ARIC) study. Circulation 1997, 96, 4219–4225. [Google Scholar] [CrossRef]
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The role of asymmetric dimethylarginine (ADMA) in endothelial dysfunction and cardiovascular disease. Curr. Cardiol. Rev. 2010, 6, 82–90. [Google Scholar] [CrossRef]
- Boger, R.H.; Maas, R.; Schulze, F.; Schwedhelm, E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality--an update on patient populations with a wide range of cardiovascular risk. Pharmacol. Res. 2009, 60, 481–487. [Google Scholar] [CrossRef]
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef]
- Samarasekera, E.J.; Neilson, J.M.; Warren, R.B.; Parnham, J.; Smith, C.H. Incidence of cardiovascular disease in individuals with psoriasis: a systematic review and meta-analysis. J. Investig. Dermatol. 2013, 133, 2340–2346. [Google Scholar]
- Crowson, C.S.; Liao, K.P.; Davis, J.M., 3rd; Solomon, D.H.; Matteson, E.L.; Knutson, K.L.; Hlatky, M.A.; Gabriel, S.E. Rheumatoid arthritis and cardiovascular disease. Am. Heart J. 2013, 166, 622–628 e621. [Google Scholar] [CrossRef]
- Solomon, D.H.; Goodson, N.J.; Katz, J.N.; Weinblatt, M.E.; Avorn, J.; Setoguchi, S.; Canning, C.; Schneeweiss, S. Patterns of cardiovascular risk in rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 1608–1612. [Google Scholar] [CrossRef]
- Avina-Zubieta, J.A.; Choi, H.K.; Sadatsafavi, M.; Etminan, M.; Esdaile, J.M.; Lacaille, D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Rheum. 2008, 59, 1690–1697. [Google Scholar] [CrossRef]
- Hak, A.E.; Karlson, E.W.; Feskanich, D.; Stampfer, M.J.; Costenbader, K.H. Systemic lupus erythematosus and the risk of cardiovascular disease: results from the nurses’ health study. Arthritis Rheum. 2009, 61, 1396–1402. [Google Scholar] [CrossRef]
- Magder, L.S.; Petri, M. Incidence of and risk factors for adverse cardiovascular events among patients with systemic lupus erythematosus. Am. J. Epidemiol. 2012, 176, 708–719. [Google Scholar] [CrossRef]
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef]
- Mathieu, S.; Gossec, L.; Dougados, M.; Soubrier, M. Cardiovascular profile in ankylosing spondylitis: A systematic review and meta-analysis. Arthritis Care Res. (Hoboken) 2011, 63, 557–563. [Google Scholar] [CrossRef]
- Peters, M.J.; Visman, I.; Nielen, M.M.; van Dillen, N.; Verheij, R.A.; van der Horst-Bruinsma, I.E.; Dijkmans, B.A.; Nurmohamed, M.T. Ankylosing spondylitis: A risk factor for myocardial infarction? Ann. Rheum. Dis. 2010, 69, 579–581. [Google Scholar] [CrossRef]
- Fumery, M.; Xiaocang, C.; Dauchet, L.; Gower-Rousseau, C.; Peyrin-Biroulet, L.; Colombel, J.F. Thromboembolic events and cardiovascular mortality in inflammatory bowel diseases: A meta-analysis of observational studies. J. Crohns Colitis 2014, 8, 469–479. [Google Scholar]
- Dorn, S.D.; Sandler, R.S. Inflammatory bowel disease is not a risk factor for cardiovascular disease mortality: Results from a systematic review and meta-analysis. Am. J. Gastroenterol. 2007, 102, 662–667. [Google Scholar] [CrossRef]
- Bergholm, R. Impaired responsiveness to NO in newly diagnosed patients with rheumatoid arthritis. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1637–1641. [Google Scholar] [CrossRef]
- Chatterjee Adhikari, M.; Guin, A.; Chakraborty, S.; Sinhamahapatra, P.; Ghosh, A. Subclinical atherosclerosis and endothelial dysfunction in patients with early rheumatoid arthritis as evidenced by measurement of carotid intima-media thickness and flow-mediated vasodilatation: An observational study. Semin. Arthritis Rheum. 2012, 41, 669–675. [Google Scholar] [CrossRef]
- Kerekes, G.; Szekanecz, Z.; Der, H.; Sandor, Z.; Lakos, G.; Muszbek, L.; Csipo, I.; Sipka, S.; Seres, I.; Paragh, G.; et al. Endothelial dysfunction and atherosclerosis in rheumatoid arthritis: A multiparametric analysis using imaging techniques and laboratory markers of inflammation and autoimmunity. J. Rheumatol. 2008, 35, 398–406. [Google Scholar]
- Vaudo, G. Endothelial dysfunction in young patients with rheumatoid arthritis and low disease activity. Ann. Rheum. Dis. 2004, 63, 31–35. [Google Scholar] [CrossRef]
- Hänsel, S.; Lässig, G.; Pistrosch, F.; Passauer, J. Endothelial dysfunction in young patients with long-term rheumatoid arthritis and low disease activity. Atherosclerosis 2003, 170, 177–180. [Google Scholar] [CrossRef]
- Syngle, A.; Vohra, K.; Kaur, L.; Sharma, S. Effect of spironolactone on endothelial dysfunction in rheumatoid arthritis. Scand. J. Rheumatol. 2009, 38, 15–22. [Google Scholar] [CrossRef]
- Datta, D.; Ferrell, W.R.; Sturrock, R.D.; Jadhav, S.T.; Sattar, N. Inflammatory suppression rapidly attenuates microvascular dysfunction in rheumatoid arthritis. Atherosclerosis 2007, 192, 391–395. [Google Scholar] [CrossRef]
- Galarraga, B.; Khan, F.; Kumar, P.; Pullar, T.; Belch, J.J. C-reactive protein: The underlying cause of microvascular dysfunction in rheumatoid arthritis. Rheumatology (Oxford) 2008, 47, 1780–1784. [Google Scholar] [CrossRef]
- Sodergren, A.; Karp, K.; Boman, K.; Eriksson, C.; Lundstrom, E.; Smedby, T.; Soderlund, L.; Rantapaa-Dahlqvist, S.; Wallberg-Jonsson, S. Atherosclerosis in early rheumatoid arthritis: very early endothelial activation and rapid progression of intima media thickness. Arthritis Res. Ther. 2010, 12, R158. [Google Scholar] [CrossRef]
- Foster, W.; Lip, G.Y.; Raza, K.; Carruthers, D.; Blann, A.D. An observational study of endothelial function in early arthritis. Eur. J. Clin. Investig. 2012, 42, 510–516. [Google Scholar] [CrossRef]
- Sandoo, A.; Veldhuijzen van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. Vascular function and morphology in rheumatoid arthritis: A systematic review. Rheumatology (Oxford) 2011, 50, 2125–2139. [Google Scholar] [CrossRef]
- Littler, A.J.; Buckley, C.D.; Wordsworth, P.; Collins, I.; Martinson, J.; Simmons, D.L. A distinct profile of six soluble adhesion molecules (ICAM-1, ICAM-3, VCAM-1, E-selectin, L-selectin and P-selectin) in rheumatoid arthritis. Br. J. Rheumatol. 1997, 36, 164–169. [Google Scholar] [CrossRef]
- Klimiuk, P.A.; Fiedorczyk, M.; Sierakowski, S.; Chwiecko, J. Soluble cell adhesion molecules (sICAM-1, sVCAM-1, and sE-selectin) in patients with early rheumatoid arthritis. Scand. J. Rheumatol. 2007, 36, 345–350. [Google Scholar] [CrossRef]
- Dessein, P.H.; Joffe, B.I.; Singh, S. Biomarkers of endothelial dysfunction, cardiovascular risk factors and atherosclerosis in rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R634–R643. [Google Scholar] [CrossRef]
- Veale, D.J.; Maple, C.; Kirk, G.; McLaren, M.; Belch, J.J. Soluble cell adhesion molecules—P-selectin and ICAM-1, and disease activity in patients receiving sulphasalazine for active rheumatoid arthritis. Scand. J. Rheumatol. 1998, 27, 296–299. [Google Scholar] [CrossRef]
- Meyer, M.F.; Schmidt, O.; Hellmich, B.; Schatz, H.; Klein, H.H.; Braun, J. Microvascular dysfunction in rheumatoid arthritis assessed by laser Doppler anemometry: Relationship to soluble adhesion molecules and extraarticular manifestations. Rheumatol. Int. 2007, 28, 145–152. [Google Scholar] [CrossRef]
- Antoniades, C.; Demosthenous, M.; Tousoulis, D.; Antonopoulos, A.S.; Vlachopoulos, C.; Toutouza, M.; Marinou, K.; Bakogiannis, C.; Mavragani, K.; Lazaros, G.; et al. Role of asymmetrical dimethylarginine in inflammation-induced endothelial dysfunction in human atherosclerosis. Hypertension 2011, 58, 93–98. [Google Scholar] [CrossRef]
- Lima, D.S.; Sato, E.I.; Lima, V.C.; Miranda, F., Jr.; Hatta, F.H. Brachial endothelial function is impaired in patients with systemic lupus erythematosus. J. Rheumatol. 2002, 29, 292–297. [Google Scholar]
- Kiss, E.; Soltesz, P.; Der, H.; Kocsis, Z.; Tarr, T.; Bhattoa, H.; Shoenfeld, Y.; Szegedi, G. Reduced flow-mediated vasodilation as a marker for cardiovascular complications in lupus patients. J. Autoimmun. 2006, 27, 211–217. [Google Scholar] [CrossRef]
- El-Magadmi, M.; Bodill, H.; Ahmad, Y.; Durrington, P.N.; Mackness, M.; Walker, M.; Bernstein, R.M.; Bruce, I.N. Systemic lupus erythematosus: An independent risk factor for endothelial dysfunction in women. Circulation 2004, 110, 399–404. [Google Scholar] [CrossRef]
- Piper, M.K.; Raza, K.; Nuttall, S.L.; Stevens, R.; Toescu, V.; Heaton, S.; Gardner-Medwin, J.; Hiller, L.; Martin, U.; Townend, J.; et al. Impaired endothelial function in systemic lupus erythematosus. Lupus 2007, 16, 84–88. [Google Scholar] [CrossRef]
- Wright, S.A.; O’Prey, F.M.; Rea, D.J.; Plumb, R.D.; Gamble, A.J.; Leahey, W.J.; Devine, A.B.; McGivern, R.C.; Johnston, D.G.; Finch, M.B.; et al. Microcirculatory hemodynamics and endothelial dysfunction in systemic lupus erythematosus. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2281–2287. [Google Scholar] [CrossRef]
- Svenungsson, E.; Cederholm, A.; Jensen-Urstad, K.; Fei, G.Z.; de Faire, U.; Frostegard, J. Endothelial function and markers of endothelial activation in relation to cardiovascular disease in systemic lupus erythematosus. Scand. J. Rheumatol. 2008, 37, 352–359. [Google Scholar] [CrossRef]
- Sfikakis, P.P.; Charalambopoulos, D.; Vayiopoulos, G.; Oglesby, R.; Sfikakis, P.; Tsokos, G.C. Increased levels of intercellular adhesion molecule-1 in the serum of patients with systemic lupus erythematosus. Clin. Exp. Rheumatol. 1994, 12, 5–9. [Google Scholar]
- Tulek, N.; Aydintug, O.; Ozoran, K.; Tutkak, H.; Duzgun, N.; Duman, M.; Tokgoz, G. Soluble intercellular adhesion molecule-1 (sICAM-1) in patients with systemic lupus erythematosus. Clin. Rheumatol. 1996, 15, 47–50. [Google Scholar] [CrossRef]
- Machold, K.P.; Kiener, H.P.; Graninger, W.; Graninger, W.B. Soluble intercellular adhesion molecule-1 (sICAM-1) in patients with rheumatoid arthritis and systemic lupus erythematosus. Clin. Immunol. Immunopathol. 1993, 68, 74–78. [Google Scholar] [CrossRef]
- Janssen, B.A.; Luqmani, R.A.; Gordon, C.; Hemingway, I.H.; Bacon, P.A.; Gearing, A.J.; Emery, P. Correlation of blood levels of soluble vascular cell adhesion molecule-1 with disease activity in systemic lupus erythematosus and vasculitis. Br. J. Rheumatol. 1994, 33, 1112–1116. [Google Scholar] [CrossRef]
- Spronk, P.E.; Bootsma, H.; Huitema, M.G.; Limburg, P.C.; Kallenberg, C.G. Levels of soluble VCAM-1, soluble ICAM-1, and soluble E-selectin during disease exacerbations in patients with systemic lupus erythematosus (SLE); a long term prospective study. Clin. Exp. Immunol. 1994, 97, 439–444. [Google Scholar]
- Sari, R.; Taysi, S.; Erdem, F.; Yilmaz, Ö.; Keleş, S.; Kiziltunç, A.; Odabaş, A.; Çetinkaya, R. Correlation of serum levels of soluble intercellular adhesion molecule-1 with disease activity in systemic lupus erythematosus. Rheumatol. Int. 2001, 21, 149–152. [Google Scholar]
- Sari, I.; Okan, T.; Akar, S.; Cece, H.; Altay, C.; Secil, M.; Birlik, M.; Onen, F.; Akkoc, N. Impaired endothelial function in patients with ankylosing spondylitis. Rheumatology (Oxford) 2006, 45, 283–286. [Google Scholar]
- Gonzalez-Juanatey, C.; Llorca, J.; Miranda-Filloy, J.A.; Amigo-Diaz, E.; Testa, A.; Garcia-Porrua, C.; Martin, J.; Gonzalez-Gay, M.A. Endothelial dysfunction in psoriatic arthritis patients without clinically evident cardiovascular disease or classic atherosclerosis risk factors. Arthritis Rheum. 2007, 57, 287–293. [Google Scholar]
- Bodnar, N.; Kerekes, G.; Seres, I.; Paragh, G.; Kappelmayer, J.; Nemethne, Z.G.; Szegedi, G.; Shoenfeld, Y.; Sipka, S.; Soltesz, P.; et al. Assessment of subclinical vascular disease associated with ankylosing spondylitis. J. Rheumatol. 2011, 38, 723–729. [Google Scholar] [CrossRef]
- Wendling, D.; Racadot, E.; Auge, B.; Toussirot, E. Soluble intercellular adhesion molecule 1 in spondylarthropathies. Clin. Rheumatol. 1998, 17, 202–204. [Google Scholar] [CrossRef]
- Sari, I.; Alacacioglu, A.; Kebapcilar, L.; Taylan, A.; Bilgir, O.; Yildiz, Y.; Yuksel, A.; Kozaci, D.L. Assessment of soluble cell adhesion molecules and soluble CD40 ligand levels in ankylosing spondylitis. Jt. Bone Spine 2010, 77, 85–87. [Google Scholar] [CrossRef]
- Kemeny-Beke, A.; Gesztelyi, R.; Bodnar, N.; Zsuga, J.; Kerekes, G.; Zsuga, M.; Biri, B.; Keki, S.; Szodoray, P.; Berta, A.; et al. Increased production of asymmetric dimethylarginine (ADMA) in ankylosing spondylitis: association with other clinical and laboratory parameters. Jt. Bone Spine 2011, 78, 184–187. [Google Scholar] [CrossRef]
- Erre, G.L.; Sanna, P.; Zinellu, A.; Ponchietti, A.; Fenu, P.; Sotgia, S.; Carru, C.; Ganau, A.; Passiu, G. Plasma asymmetric dimethylarginine (ADMA) levels and atherosclerotic disease in ankylosing spondylitis: A cross-sectional study. Clin. Rheumatol. 2011, 30, 21–27. [Google Scholar] [CrossRef]
- Brezinski, E.A.; Follansbee, M.R.; Armstrong, E.J.; Armstrong, A.W. Endothelial dysfunction and the effects of TNF inhibitors on the endothelium in psoriasis and psoriatic arthritis: A systematic review. Curr. Pharm. Des. 2014, 20, 513–528. [Google Scholar] [CrossRef]
- Balci, D.D.; Balci, A.; Karazincir, S.; Ucar, E.; Iyigun, U.; Yalcin, F.; Seyfeli, E.; Inandi, T.; Egilmez, E. Increased carotid artery intima-media thickness and impaired endothelial function in psoriasis. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 1–6. [Google Scholar]
- Usta, M.; Yurdakul, S.; Aral, H.; Turan, E.; Oner, E.; Inal, B.B.; Oner, F.A.; Gurel, M.S.; Guvenen, G. Vascular endothelial function assessed by a noninvasive ultrasound method and serum asymmetric dimethylarginine concentrations in mild-to-moderate plaque-type psoriatic patients. Clin. Biochem. 2011, 44, 1080–1084. [Google Scholar] [CrossRef]
- Martyn-Simmons, C.L.; Ranawaka, R.R.; Chowienczyk, P.; Crook, M.A.; Marber, M.S.; Smith, C.H.; Barker, J.N. A prospective case-controlled cohort study of endothelial function in patients with moderate to severe psoriasis. Br. J. Dermatol. 2011, 164, 26–32. [Google Scholar] [CrossRef]
- Hatoum, O.A.; Binion, D.G.; Otterson, M.F.; Gutterman, D.D. Acquired microvascular dysfunction in inflammatory bowel disease: Loss of nitric oxide-mediated vasodilation. Gastroenterology 2003, 125, 58–69. [Google Scholar] [CrossRef]
- Kocaman, O.; Sahin, T.; Aygun, C.; Senturk, O.; Hulagu, S. Endothelial dysfunction in patients with ulcerative colitis. Inflamm. Bowel Dis. 2006, 12, 166–171. [Google Scholar] [CrossRef]
- Principi, M.; Mastrolonardo, M.; Scicchitano, P.; Gesualdo, M.; Sassara, M.; Guida, P.; Bucci, A.; Zito, A.; Caputo, P.; Albano, F.; et al. Endothelial function and cardiovascular risk in active inflammatory bowel diseases. J. Crohns Colitis 2013, 7, e427–e433. [Google Scholar] [CrossRef]
- Aloi, M.; Tromba, L.; di Nardo, G.; Dilillo, A.; del Giudice, E.; Marocchi, E.; Viola, F.; Civitelli, F.; Berni, A.; Cucchiara, S. Premature subclinical atherosclerosis in pediatric inflammatory bowel disease. J. Pediatr. 2012, 161, 589–594 e581. [Google Scholar] [CrossRef]
- Brand, D.D.; Latham, K.A.; Rosloniec, E.F. Collagen-induced arthritis. Nat. Protoc. 2007, 2, 1269–1275. [Google Scholar] [CrossRef]
- He, M.; Liang, X.; He, L.; Wen, W.; Zhao, S.; Wen, L.; Liu, Y.; Shyy, J.Y.; Yuan, Z. Endothelial dysfunction in rheumatoid arthritis: The role of monocyte chemotactic protein-1-induced protein. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1384–1391. [Google Scholar] [CrossRef]
- Denys, A.; Clavel, G.; Semerano, L.; Lemeiter, D.; Boissier, M.C. A1.6 Vascular adhesion molecule VCAM-1 overexpression in collagen induced arthritis: A model for rheumatoid arthritis vascular dysfunction. Ann. Rheum. Dis. 2014, 73, A3. [Google Scholar]
- Haruna, Y.; Morita, Y.; Komai, N.; Yada, T.; Sakuta, T.; Tomita, N.; Fox, D.A.; Kashihara, N. Endothelial dysfunction in rat adjuvant-induced arthritis: Vascular superoxide production by NAD(P)H oxidase and uncoupled endothelial nitric oxide synthase. Arthritis Rheum. 2006, 54, 1847–1855. [Google Scholar] [CrossRef]
- Fang, Z.Y.; Fontaine, J.; Unger, P.; Berkenboom, G. Alterations of the endothelial function of isolated aortae in rats with adjuvant arthritis. Arch. Int. Pharmacodyn. Ther. 1991, 311, 122–130. [Google Scholar]
- Dooley, L.M.; Washington, E.A.; Abdalmula, A.; Tudor, E.M.; Kimpton, W.G.; Bailey, S.R. Endothelial dysfunction in an ovine model of collagen-induced arthritis. J. Vasc. Res. 2014, 51, 90–101. [Google Scholar]
- Neumann, P.; Gertzberg, N.; Johnson, A. TNF-alpha induces a decrease in eNOS promoter activity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L452–L459. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Heusch, G.; Schulz, R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol. Ther. 2010, 127, 295–314. [Google Scholar] [CrossRef]
- Rajan, S.; Ye, J.; Bai, S.; Huang, F.; Guo, Y.L. NF-kappaB, but not p38 MAP kinase, is required for TNF-alpha-induced expression of cell adhesion molecules in endothelial cells. J. Cell. Biochem. 2008, 105, 477–486. [Google Scholar] [CrossRef]
- Bergh, N.; Ulfhammer, E.; Glise, K.; Jern, S.; Karlsson, L. Influence of TNF-alpha and biomechanical stress on endothelial anti- and prothrombotic genes. Biochem. Biophys. Res. Commun. 2009, 385, 314–318. [Google Scholar] [CrossRef]
- Wang, P.; Ba, Z.F.; Chaudry, I.H. Administration of tumor necrosis factor-alpha in vivo depresses endothelium-dependent relaxation. Am. J. Physiol. 1994, 266, H2535–H2541. [Google Scholar]
- Chia, S.; Qadan, M.; Newton, R.; Ludlam, C.A.; Fox, K.A.; Newby, D.E. Intra-arterial tumor necrosis factor-alpha impairs endothelium-dependent vasodilatation and stimulates local tissue plasminogen activator release in humans. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 695–701. [Google Scholar] [CrossRef]
- Hingorani, A.D.; Cross, J.; Kharbanda, R.K.; Mullen, M.J.; Bhagat, K.; Taylor, M.; Donald, A.E.; Palacios, M.; Griffin, G.E.; Deanfield, J.E.; et al. Acute systemic inflammation impairs endothelium-dependent dilatation in humans. Circulation 2000, 102, 994–999. [Google Scholar] [CrossRef]
- Ohta, H.; Wada, H.; Niwa, T.; Kirii, H.; Iwamoto, N.; Fujii, H.; Saito, K.; Sekikawa, K.; Seishima, M. Disruption of tumor necrosis factor-alpha gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis 2005, 180, 11–17. [Google Scholar] [CrossRef]
- Kundu, S.; Ghosh, P.; Datta, S.; Ghosh, A.; Chattopadhyay, S.; Chatterjee, M. Oxidative stress as a potential biomarker for determining disease activity in patients with rheumatoid arthritis. Free Radic. Res. 2012, 46, 1482–1489. [Google Scholar] [CrossRef]
- Picchi, A.; Gao, X.; Belmadani, S.; Potter, B.J.; Focardi, M.; Chilian, W.M.; Zhang, C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ. Res. 2006, 99, 69–77. [Google Scholar] [CrossRef]
- Kalinowski, L.; Malinski, T. Endothelial NADH/NADPH-dependent enzymatic sources of superoxide production: Relationship to endothelial dysfunction. Acta Biochim. Pol. 2004, 51, 459–469. [Google Scholar]
- Gielis, J.F.; Lin, J.Y.; Wingler, K.; van Schil, P.E.; Schmidt, H.H.; Moens, A.L. Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic. Biol. Med. 2011, 50, 765–776. [Google Scholar] [CrossRef]
- Hamilton, C.A.; Miller, W.H.; Al-Benna, S.; Brosnan, M.J.; Drummond, R.D.; McBride, M.W.; Dominiczak, A.F. Strategies to reduce oxidative stress in cardiovascular disease. Clin. Sci. (Lond.) 2004, 106, 219–234. [Google Scholar] [CrossRef]
- Wolin, M.S. Interactions of oxidants with vascular signaling Systems. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1430–1442. [Google Scholar] [CrossRef]
- Biniecka, M.; Kennedy, A.; Ng, C.T.; Chang, T.C.; Balogh, E.; Fox, E.; Veale, D.J.; Fearon, U.; O’Sullivan, J.N. Successful tumour necrosis factor (TNF) blocking therapy suppresses oxidative stress and hypoxia-induced mitochondrial mutagenesis in inflammatory arthritis. Arthritis Res. Ther. 2011, 13, R121. [Google Scholar] [CrossRef]
- Ku, I.A.; Imboden, J.B.; Hsue, P.Y.; Ganz, P. Rheumatoid arthritis: Model of systemic inflammation driving atherosclerosis. Circ. J. 2009, 73, 977–985. [Google Scholar] [CrossRef]
- Hurt-Camejo, E.; Paredes, S.; Masana, L.; Camejo, G.; Sartipy, P.; Rosengren, B.; Pedreno, J.; Vallve, J.C.; Benito, P.; Wiklund, O. Elevated levels of small, low-density lipoprotein with high affinity for arterial matrix components in patients with rheumatoid arthritis: Possible contribution of phospholipase A2 to this atherogenic profile. Arthritis Rheum. 2001, 44, 2761–2767. [Google Scholar] [CrossRef]
- Norata, G.D.; Tonti, L.; Roma, P.; Catapano, A.L. Apoptosis and proliferation of endothelial cells in early atherosclerotic lesions: possible role of oxidised LDL. Nutr. Metab. Cardiovasc. Dis. 2002, 12, 297–305. [Google Scholar]
- Vladimirova-Kitova, L.; Deneva, T.; Angelova, E.; Nikolov, F.; Marinov, B.; Mateva, N. Relationship of asymmetric dimethylarginine with flow-mediated dilatation in subjects with newly detected severe hypercholesterolemia. Clin. Physiol. Funct. Imaging 2008, 28, 417–425. [Google Scholar] [CrossRef]
- Stancu, C.S.; Toma, L.; Sima, A.V. Dual role of lipoproteins in endothelial cell dysfunction in atherosclerosis. Cell. Tissue Res. 2012, 349, 433–446. [Google Scholar] [CrossRef]
- Narshi, C.B.; Giles, I.P.; Rahman, A. The endothelium: an interface between autoimmunity and atherosclerosis in systemic lupus erythematosus? Lupus 2011, 20, 5–13. [Google Scholar] [CrossRef]
- Cieslik, P.; Hrycek, A.; Klucinski, P. Vasculopathy and vasculitis in systemic lupus erythematosus. Pol. Arch. Med. Wewn. 2008, 118, 57–63. [Google Scholar]
- Gomez-Zumaquero, J.M.; Tinahones, F.J.; de Ramon, E.; Camps, M.; Garrido, L.; Soriguer, F.J. Association of biological markers of activity of systemic lupus erythematosus with levels of anti-oxidized low-density lipoprotein antibodies. Rheumatology (Oxford) 2004, 43, 510–513. [Google Scholar] [CrossRef]
- Charakida, M.; Besler, C.; Batuca, J.R.; Sangle, S.; Marques, S.; Sousa, M.; Wang, G.; Tousoulis, D.; Delgado Alves, J.; Loukogeorgakis, S.P.; et al. Vascular abnormalities, paraoxonase activity, and dysfunctional HDL in primary antiphospholipid syndrome. JAMA 2009, 302, 1210–1217. [Google Scholar] [CrossRef]
- Hjeltnes, G.; Hollan, I.; Forre, O.; Wiik, A.; Lyberg, T.; Mikkelsen, K.; Agewall, S. Endothelial function improves within 6 weeks of treatment with methotrexate or methotrexate in combination with a TNF-alpha inhibitor in rheumatoid arthritis patients. Scand. J. Rheumatol. 2012, 41, 240–242. [Google Scholar] [CrossRef]
- Cardillo, C.; Schinzari, F.; Mores, N.; Mettimano, M.; Melina, D.; Zoli, A.; Ferraccioli, G. Intravascular tumor necrosis factor alpha blockade reverses endothelial dysfunction in rheumatoid arthritis. Clin. Pharmacol. Ther. 2006, 80, 275–281. [Google Scholar] [CrossRef]
- Bilsborough, W.; Keen, H.; Taylor, A.; O’Driscoll, G.J.; Arnolda, L.; Green, D.J. Anti-tumour necrosis factor-alpha therapy over conventional therapy improves endothelial function in adults with rheumatoid arthritis. Rheumatol. Int. 2006, 26, 1125–1131. [Google Scholar] [CrossRef]
- Hurlimann, D. Anti-tumor necrosis factor-alpha treatment improves endothelial function in patients with rheumatoid arthritis. Circulation 2002, 106, 2184–2187. [Google Scholar] [CrossRef]
- Gonzalez-Juanatey, C.; Testa, A.; Garcia-Castelo, A.; Garcia-Porrua, C.; Llorca, J.; Gonzalez-Gay, M.A. Active but transient improvement of endothelial function in rheumatoid arthritis patients undergoing long-term treatment with anti-tumor necrosis factor alpha antibody. Arthritis Rheum. 2004, 51, 447–450. [Google Scholar] [CrossRef]
- Sidiropoulos, P.I.; Siakka, P.; Pagonidis, K.; Raptopoulou, A.; Kritikos, H.; Tsetis, D.; Boumpas, D.T. Sustained improvement of vascular endothelial function during anti-TNFalpha treatment in rheumatoid arthritis patients. Scand. J. Rheumatol. 2009, 38, 6–10. [Google Scholar] [CrossRef]
- Bosello, S.; Santoliquido, A.; Zoli, A.; di Campli, C.; Flore, R.; Tondi, P.; Ferraccioli, G. TNF-alpha blockade induces a reversible but transient effect on endothelial dysfunction in patients with long-standing severe rheumatoid arthritis. Clin. Rheumatol. 2008, 27, 833–839. [Google Scholar] [CrossRef]
- Komai, N.; Morita, Y.; Sakuta, T.; Kuwabara, A.; Kashihara, N. Anti-tumor necrosis factor therapy increases serum adiponectin levels with the improvement of endothelial dysfunction in patients with rheumatoid arthritis. Mod. Rheumatol. 2007, 17, 385–390. [Google Scholar] [CrossRef]
- Van Eijk, I.C.; Peters, M.J.; Serne, E.H.; van der Horst-Bruinsma, I.E.; Dijkmans, B.A.; Smulders, Y.M.; Nurmohamed, M.T. Microvascular function is impaired in ankylosing spondylitis and improves after tumour necrosis factor alpha blockade. Ann. Rheum. Dis. 2009, 68, 362–366. [Google Scholar]
- Syngle, A.; Vohra, K.; Sharma, A.; Kaur, L. Endothelial dysfunction in ankylosing spondylitis improves after tumor necrosis factor-alpha blockade. Clin. Rheumatol. 2010, 29, 763–770. [Google Scholar] [CrossRef]
- Avgerinou, G.; Tousoulis, D.; Siasos, G.; Oikonomou, E.; Maniatis, K.; Papageorgiou, N.; Paraskevopoulos, T.; Miliou, A.; Koumaki, D.; Latsios, G.; et al. Anti-tumor necrosis factor alpha treatment with adalimumab improves significantly endothelial function and decreases inflammatory process in patients with chronic psoriasis. Int. J. Cardiol. 2011, 151, 382–383. [Google Scholar] [CrossRef]
- Schinzari, F.; Armuzzi, A.; de Pascalis, B.; Mores, N.; Tesauro, M.; Melina, D.; Cardillo, C. Tumor necrosis factor-alpha antagonism improves endothelial dysfunction in patients with Crohn’s disease. Clin. Pharmacol. Ther. 2008, 83, 70–76. [Google Scholar] [CrossRef]
- Galarraga, B.; Belch, J.J.; Pullar, T.; Ogston, S.; Khan, F. Clinical improvement in rheumatoid arthritis is associated with healthier microvascular function in patients who respond to antirheumatic therapy. J. Rheumatol. 2010, 37, 521–528. [Google Scholar] [CrossRef]
- Kerekes, G.; Soltesz, P.; Der, H.; Veres, K.; Szabo, Z.; Vegvari, A.; Szegedi, G.; Shoenfeld, Y.; Szekanecz, Z. Effects of rituximab treatment on endothelial dysfunction, carotid atherosclerosis, and lipid profile in rheumatoid arthritis. Clin. Rheumatol. 2009, 28, 705–710. [Google Scholar] [CrossRef]
- Veselinovic, M.V.; Zivkovic, V.I.; Toncev, S.; Tasic, N.; Bogdanovic, V.; Djuric, D.M.; Jakovljevic, V. Carotid artery intima-media thickness and brachial artery flow-mediated vasodilatation in patients with rheumatoid arthritis. VASA 2012, 41, 343–351. [Google Scholar] [CrossRef]
- Hafstrom, I.; Rohani, M.; Deneberg, S.; Wornert, M.; Jogestrand, T.; Frostegard, J. Effects of low-dose prednisolone on endothelial function, atherosclerosis, and traditional risk factors for atherosclerosis in patients with rheumatoid arthritis--a randomized study. J. Rheumatol. 2007, 34, 1810–1816. [Google Scholar]
- Maki-Petaja, K.M.; Booth, A.D.; Hall, F.C.; Wallace, S.M.; Brown, J.; McEniery, C.M.; Wilkinson, I.B. Ezetimibe and simvastatin reduce inflammation, disease activity, and aortic stiffness and improve endothelial function in rheumatoid arthritis. J. Am. Coll. Cardiol. 2007, 50, 852–858. [Google Scholar] [CrossRef]
- Tikiz, C.; Utuk, O.; Pirildar, T.; Bayturan, O.; Bayindir, P.; Taneli, F.; Tikiz, H.; Tuzun, C. Effects of Angiotensin-converting enzyme inhibition and statin treatment on inflammatory markers and endothelial functions in patients with longterm rheumatoid arthritis. J. Rheumatol. 2005, 32, 2095–2101. [Google Scholar]
- Hermann, F.; Forster, A.; Chenevard, R.; Enseleit, F.; Hurlimann, D.; Corti, R.; Spieker, L.E.; Frey, D.; Hermann, M.; Riesen, W.; et al. Simvastatin improves endothelial function in patients with rheumatoid arthritis. J. Am. Coll. Cardiol. 2005, 45, 461–464. [Google Scholar] [CrossRef]
- Ferreira, G.A.; Navarro, T.P.; Telles, R.W.; Andrade, L.E.; Sato, E.I. Atorvastatin therapy improves endothelial-dependent vasodilation in patients with systemic lupus erythematosus: An 8 weeks controlled trial. Rheumatology (Oxford) 2007, 46, 1560–1565. [Google Scholar] [CrossRef]
- Klimiuk, P.A.; Sierakowski, S.; Domyslawska, I.; Chwiecko, J. Effect of etanercept on serum levels of soluble cell adhesion molecules (sICAM-1, sVCAM-1, and sE-selectin) and vascular endothelial growth factor in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2009, 38, 439–444. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Garcia-Unzueta, M.T.; de Matias, J.M.; Gonzalez-Juanatey, C.; Garcia-Porrua, C.; Sanchez-Andrade, A.; Martin, J.; Llorca, J. Influence of anti-TNF-alpha infliximab therapy on adhesion molecules associated with atherogenesis in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2006, 24, 373–379. [Google Scholar]
- Ruyssen-Witrand, A.; Fautrel, B.; Saraux, A.; Le Loet, X.; Pham, T. Cardiovascular risk induced by low-dose corticosteroids in rheumatoid arthritis: A systematic literature review. Jt. Bone Spine 2011, 78, 23–30. [Google Scholar] [CrossRef]
- Roman, M.J.; Shanker, B.A.; Davis, A.; Lockshin, M.D.; Sammaritano, L.; Simantov, R.; Crow, M.K.; Schwartz, J.E.; Paget, S.A.; Devereux, R.B.; et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 2399–2406. [Google Scholar] [CrossRef]
- Palinski, W. Unraveling pleiotropic effects of statins on plaque rupture. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1745–1750. [Google Scholar] [CrossRef]
- Kanda, H.; Hamasaki, K.; Kubo, K.; Tateishi, S.; Yonezumi, A.; Kanda, Y.; Yamamoto, K.; Mimura, T. Antiinflammatory effect of simvastatin in patients with rheumatoid arthritis. J. Rheumatol. 2002, 29, 2024–2026. [Google Scholar]
- McCarey, D.W.; McInnes, I.B.; Madhok, R.; Hampson, R.; Scherbakova, O.; Ford, I.; Capell, H.A.; Sattar, N. Trial of Atorvastatin in Rheumatoid Arthritis (TARA): Double-blind, randomised placebo-controlled trial. Lancet 2004, 363, 2015–2021. [Google Scholar] [CrossRef]
- Okamoto, H.; Koizumi, K.; Kamitsuji, S.; Inoue, E.; Hara, M.; Tomatsu, T.; Kamatani, N.; Yamanaka, H. Beneficial action of statins in patients with rheumatoid arthritis in a large observational cohort. J. Rheumatol. 2007, 34, 964–968. [Google Scholar]
- Sheng, X.; Murphy, M.J.; Macdonald, T.M.; Wei, L. Effectiveness of statins on total cholesterol and cardiovascular disease and all-cause mortality in osteoarthritis and rheumatoid arthritis. J. Rheumatol. 2012, 39, 32–40. [Google Scholar] [CrossRef]
- Semb, A.G.; Kvien, T.K.; DeMicco, D.A.; Fayyad, R.; Wun, C.C.; LaRosa, J.C.; Betteridge, J.; Pedersen, T.R.; Holme, I. Effect of intensive lipid-lowering therapy on cardiovascular outcome in patients with and those without inflammatory joint disease. Arthritis Rheum. 2012, 64, 2836–2846. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).

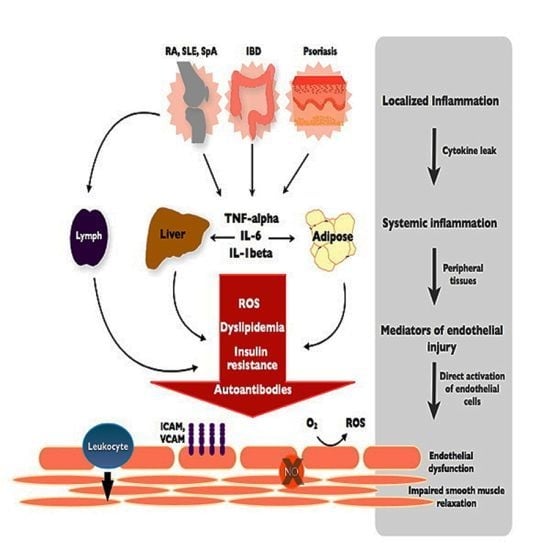

{kind=link}
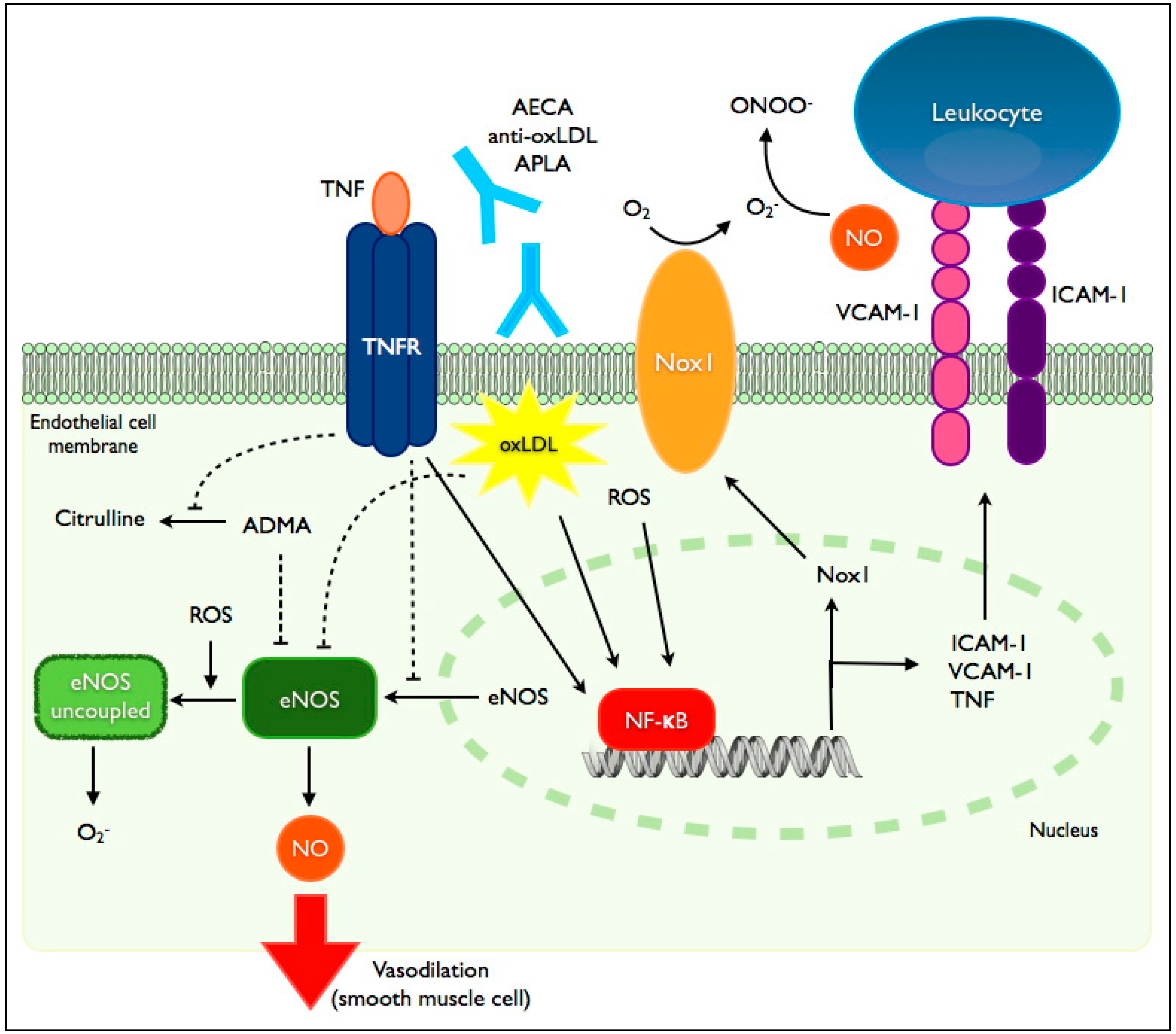{kind=link}
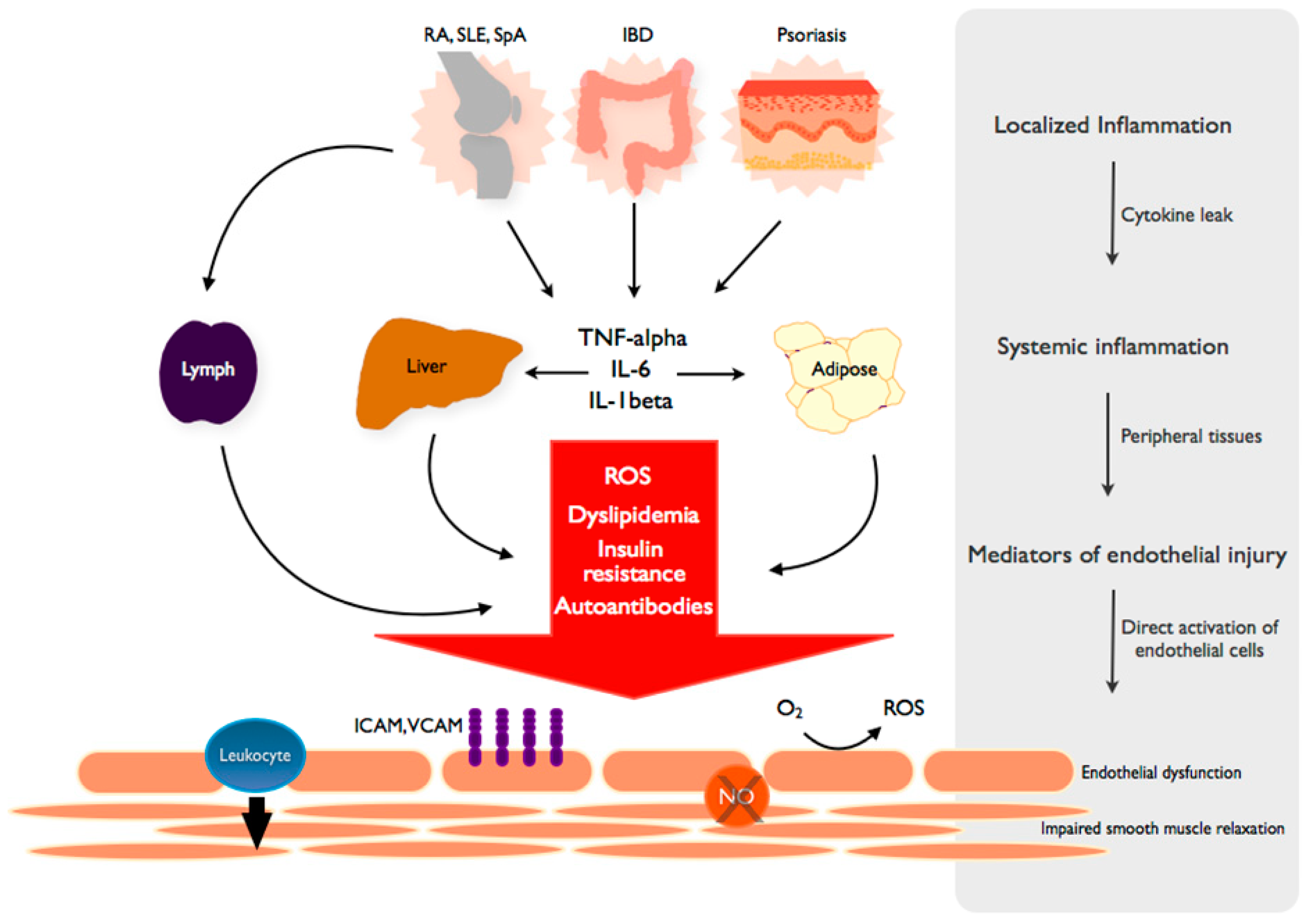{kind=link}

