HIV-1 Envelope Glycoprotein at the Interface of Host Restriction and Virus Evasion


1
Department of Microbiology & Immunology, McGill University, Montreal, QC H3A 2B4, Canada
2
Lady Davis Institute, Jewish General Hospital, Montreal, QC H3T 1E2, Canada
3
Department of Medicine, McGill University, Montreal, QC H4A 3J1, Canada
4
Center for Retrovirus Research, Department of Veterinary Biosciences, Department of Microbial Infection and Immunity, The Ohio State University, Columbus, Ohio 43210, USA
*
Author to whom correspondence should be addressed.
Viruses 2019, 11(4), 311; https://doi.org/10.3390/v11040311
Submission received: 4 March 2019
/
Revised: 25 March 2019
/
Accepted: 27 March 2019
/
Published: 30 March 2019
(This article belongs to the Special Issue Viruses Ten-Year Anniversary)
Abstract
:Without viral envelope proteins, viruses cannot enter cells to start infection. As the major viral proteins present on the surface of virions, viral envelope proteins are a prominent target of the host immune system in preventing and ultimately eliminating viral infection. In addition to the well-appreciated adaptive immunity that produces envelope protein-specific antibodies and T cell responses, recent studies have begun to unveil a rich layer of host innate immune mechanisms restricting viral entry. This review focuses on the exciting progress that has been made in this new direction of research, by discussing various known examples of host restriction of viral entry, and diverse viral countering strategies, in particular, the emerging role of viral envelope proteins in evading host innate immune suppression. We will also highlight the effective cooperation between innate and adaptive immunity to achieve the synergistic control of viral infection by targeting viral envelope protein and checking viral escape. Given that many of the related findings were made with HIV-1, we will use HIV-1 as the model virus to illustrate the basic principles and molecular mechanisms on host restriction targeting HIV-1 envelope protein.
1. Introduction
The HIV-1 envelope (Env) protein is synthesized as a gp160 precursor at the endoplasmic reticulum (ER). Following complex glycosylation and cleavage by furin proteases at the trans-Golgi complex, the mature gp120/gp41 trimer travels to the plasma membrane where it joins HIV-1 Gag proteins, forming infectious virus particles (reviewed in [1]). To start a new round of infection, gp120 binds to the CD4 receptor on the surface of the target cell, which triggers conformational changes of the gp120/gp41 trimer, exposing the binding site in gp120 for the CCR5 or the CXCR4 co-receptor. Engagement of the co-receptor leads to the exposure of the fusion peptide followed by the assembly of the 6-helix bundle in gp41, causing the fusion of viral and cellular membranes. Membrane fusion begins with the joining of both membranes’ outer lipid leaflets in a process called hemifusion. Continued fusion of the two membranes leads to the formation of the fusion pore. The fusion pore further dilates to an adequate size for the delivery of HIV-1 RNA within the core structure into the cytoplasm.
As the only viral protein on the surface of HIV-1 particles, the Env protein represents the chief target for recognition by the host adaptive immune system, leading to the production of antibodies that recognize and bind to Env protein. Despite the various strategies that HIV-1 has exploited to evade neutralizing antibodies, including heavy glycosylation to mask the epitopes [2], a series of antibodies that are able to neutralize diverse HIV-1 strains has been isolated from HIV patients, called broadly neutralizing antibodies (bnAbs) (reviewed in [3]). The discovery of bnAbs has fueled HIV vaccine research through the characterization of bnAb epitopes on the Env protein and the elucidation of the B cell generation of bnAbs [4].
In addition to neutralizing HIV-1 particles, some Env-targeting antibodies are able to recruit natural killer (NK) cells through engaging the FC receptor, and kill HIV-1 infected cells by antibody-dependent cell-mediated cytotoxicity (ADCC) [5]. The ADCC-mediating Env Abs were isolated from subjects enrolled in the RV144 vaccine trial, which has shown a modest 31.2% protection efficacy [6]. Not surprisingly, HIV-1 has mechanisms to evade ADCC, including the use of accessory proteins Vpu and Nef to downregulate CD4, which otherwise interacts with the Env protein to expose epitopes of ADCC-triggering antibodies (reviewed in [7]).
2. HIV-1 Env Is Attacked by Host Innate Immunity
In addition to the constitutively active intrinsic innate immune mechanisms, the main stream of innate immunity is elicited upon host recognition of pathogen-associated molecular patterns (PAMPs) by a group of host proteins that act as pathogen recognition receptors (PRRs). This recognition event triggers signaling cascades that lead to the expression of cytokines, including interferons (IFNs). One function of these cytokines is to induce the expression of proteins that can directly restrict viral infections. The other equally important function of cytokines is to activate immune cells and initiate the pathogen-specific adaptive immune response.
By acting together, the constitutively expressed and interferon-induced antiviral proteins form the first line of host defense against viral infections. These antiviral proteins operate by a variety of molecular mechanisms to target distinct steps in the virus life cycle. As viral nucleic acids are the main PAMPs to induce innate immune response, they have also become the target of many cellular antiviral factors (reviewed in [8]). Examples include (1) nucleases, such as ISG20, OAS/RNase L, and three primer repair exonuclease 1 (TREX1) that degrade viral RNA or DNA; (2) deaminases, such as adenosine deaminases acting on RNA (ADAR) and apoliprotein B editing complex 3 (APOBEC3) proteins that edit and mutate the viral genome; (3) dNTP hydrolase SAM domain and HD domain-containing protein 1 (SAMHD1), which diminishes the cellular DNA pool and inhibits viral DNA synthesis; (4) factors, such as Myxovirus resistance 1, 2 (Mx1, Mx2), and Tripartite motif-containing protein 5α (Trim5α) that target the replication complex of viral genome and block viral multiplication; and (5) factors such as schlafen 11 (SLFN11) and protein kinase activated by RNA (PKR), which inhibit the translation of viral RNA [9,10,11,12,13,14,15,16].
In addition to these diverse cellular mechanisms that attack viral nucleic acids, recent studies have discovered an array of cellular proteins that contribute to the control of viral infection by targeting viral Env and inhibiting viral entry, which began to unravel a new layer of host antiviral defense. Through these studies, we have come to appreciate the combinatorial strategy that cells have evolved to restrict viral entry by targeting virtually every stage of Env’s life, from Env synthesis and maturation, to its incorporation into virus particles, to its execution of membrane fusion.
3. A Long and Challenging Journey for Env Protein to Reach Virus Particles
From its de novo synthesis at the ER, HIV-1 Env protein travels through the trans-Golgi complex, arriving at the plasma membrane to join HIV-1 Gag proteins, together forming infectious progeny virions. Env can choose to detour to endosome recycle compartments (ERC), where it reaches the particle assembly site on the plasma membrane by interacting with FIP1C and Rab14 [17,18]. Along this journey, Env undergoes complex glycosylation, trimerization, and cleavage by furin, providing ample opportunities for the host cell to attack (Figure 1).
3.1. ER-Associated Degradation: Traps at Its Place of Birth
The newly synthesized Env precursor, gp160, folds at the ER with low efficiency [20]. The often misfolded Env protein is subjected to ER-associated degradation (ERAD), a process that controls and eliminates misfolded proteins [21]. One ER protein, called ERManI, has been shown to promote degradation of ER-associated proteins by ERAD. ERManI is a class I α-mannosidase and belongs to the glycosidehydrolase family 47 (GH47) α-mannosidases, which are carbohydrate-active enzymes [22]. GH47 α-mannosidases mediate the trimming of α-1,2-mannose residues from Man9GlcNAc2, among which ERManI is the first enzyme to generate Man8GlcNAc2 [23]. ERManl interacts with HIV-1 Env via its luminal catalytic domain, and mutation of the catalytic sites ablates its activity in degrading HIV-1 Env protein [24]. This function of ERManl is specific, because overexpression of other α-mannosidases from the family of GH47 α-mannosidases, such as ER-degradation enhancing α-mannosidase-like (EDEM) proteins 1, 2, and 3, does not affect HIV-1 Env expression [24]. It is thus speculated that ERManI operates by modulating glycosylation of HIV-1 Env. Interestingly, ERManI is required for the function of a mitochondrial translocator protein called TSPO in diminishing HIV-1 Env expression via ERAD since ERManI depletion abolished TSPO-mediated Env degradation [24,25].
The antiviral activity of ERManI is not limited to the degradation of HIV-1 Env protein, and the hemagglutinin (HA) glycoprotein of influenza virus is also a target of ERManI and the ERAD pathway [26]. To date, viral countermeasures of ERManI have not been reported [24]. However, HIV-1 Vpr has been shown to increase Env expression [27]. In the absence of Vpr, Env tends to misfold and is targeted to the ERAD pathway for degradation. The N-terminal region of Vpr controls this activity, since a single A30L mutation disrupts the function of Vpr to increase Env expression [27]. It remains to be determined whether Vpr directly interacts with ERManI to save Env from degradation at the ER.
The ERAD-mediated degradation of HIV-1 Env can be exploited for therapeutic purposes. One example is the depletion of HIV-1 gp160 precursor from the ER via the ERAD pathway by an engineered molecule called degradin, which contains gp120-targeting antibody chains and the C-terminal sequence of ER-resident protein SEL1L [28]. By a similar mechanism, the small peptide glycine-prolyl-glycine amide (GPG-NH2) abolishes HIV-1 infectivity by targeting viral Env to the ERAD pathway for degradation [29]. The ERAD pathway appears to be a double-edged sword, since HIV-1 Vpu hijacks this protein degradation mechanism to remove CD4 from the ER [30]. Premature contact of Env with CD4 is thus avoided to ensure that Env is safely transported to the plasma membrane for virus assembly.
3.2. GBP5, 90K, and IFITM3: Blocks Along Env’s Route to the Virus Assembly Site
The precursor of HIV-1 Env, gp160, trimerizes at the ER then moves to the trans-Golgi apparatus where it is cleaved to become gp120/gp41, forming a mature Env trimer [1]. Products of three interferon-stimulated genes (ISGs), GBP5 (guanylate binding protein 5), 90K, and IFITM2/3 (IFN-induced transmembrane protein 2 and 3), have been shown to obstruct gp160 cleavage and diminish the incorporation of functional mature gp120/gp41 trimers into HIV-1 particles [31,32,33].
GBP5 is a member of IFN-inducible guanosine triphosphatases (GTPases) [34]. Members of the GBP family have been reported to antagonize a variety of invading pathogens including viruses, bacteria, and protozoa [35]. GBP1, a protein that is closely related to GBP5, inhibits a number of viruses including dengue virus, hepatitis C virus (HCV), encephalomyocarditis virus, and vesicular stomatitis virus (VSV) [36,37]. GBP5 was identified as a potential anti-HIV-1 factor in a genome-wide study for human genes sharing evolutionary signature of known restriction factors [38]. To supplement this identification, levels of GBP5 in primary CD4+ T cells and macrophages are enhanced by IFN-α, IFN-γ, IL-2, and TCR activation [31,34,39]. Not only does ectopic expression of GBP5 reduce the infectivity of HIV-1 particles by diminishing virion incorporation of gp120/gp41, depletion of GBP5 in primary macrophages elevates HIV-1 infectivity by enhancing the incorporation of mature gp120/gp41 into virions [31]. This action of GBP5 appears specific to retroviruses, since the function of VSV glycoprotein was not affected. GBP5’s location is crucial to its inhibitory effect on HIV-1; the intact C-terminal domain responsible of localizing GBP5 in the Golgi apparatus is required in lieu of GTPase activity. Ectopically introduced GBP5 causes two defects to HIV-1 Env protein as detected with Western blotting; gp160 cleavage is impaired, and glycosylation of HIV-1 Env is altered. It is currently unclear whether this altered glycosylation has contributed to the impaired cleavage of gp160. Within the GBP family, this anti-HIV-1 function appears to be specific to GBP5, as GBP1 does not affect HIV-1 Env despite its trans-Golgi localization. HIV-1 has a “trade-off” mechanism to partially overcome GBP5 inhibition through shutting down Vpu expression. Since Vpu and Env are synthesized from a single bicistronic mRNA, shutting down Vpu expression increases Env expression, which confers partial resistance to GBP5 [40]. Interestingly, the Vpu mutation that causes a loss of Vpu expression was identified in macrophage-tropic HIV-1 and some brain-derived HIV-1 strains, indicating that HIV-1 might have been pressured to resist high levels of GBP5 in macrophages [31,41].
The 90K protein (also known as Mac-2BP or LGAL3SBP) is an IFN-inducible, secreted immunostimulatory glycoprotein; it belongs to the family of scavenger receptor cysteine-rich (SRCR) domain-containing proteins [42]. In response to IFN-α stimulation, levels of 90K increase in various T cell lines, primary CD4+ T cells, and primary macrophages. Elevated levels of 90K have been reported in HIV-1 infected patients; hence, it was proposed as a serological marker of disease progression to AIDS [43,44]. 90K is N-glycosylated in the ER and Golgi complex before entry to the secretory pathway, thereby sharing the same route of trafficking and modification with HIV-1 Env protein [45]. Ectopic expression of 90K causes an accumulation of gp160, concomitant reduction of gp120, and loss of mature gp120/gp41 in HIV-1 particles, which result in impaired infectivity of nascent HIV-1 particles [32]. In addition, knockdown of 90K with siRNA in primary macrophages increases virion-associated gp120 and HIV-1 infectivity. It is also noted that both GBP5 and 90K are highly expressed in macrophages and may contribute to the low infection of macrophages by HIV-1. 90K inhibits both R5 and X4 HIV viruses [32]. While 90K also affects the furin-dependent maturation of Ebola GP, it minimally changes the processing of influenza virus HA0 protein and cellular glypican-3, suggesting a selectivity of 90K action on furin substrates. Lastly, the inhibitory effect of 90K on HIV-1 Env might be indirect, given the lack of detectable interaction of 90K with HIV-1 Env [32].
The anti-HIV-1 activity of 90K has been further mapped to the two central protein-binding domains of BTB-POZ and IVR, whereas the N-terminal scavenger receptor cysteine rich (SRCR)-like domain is dispensable [32]. However, a mutagenesis study by a different group showed that one truncation mutant of 90K (1-95) inhibits Env processing, while another 90K mutant (124-585) inhibits virion production [46]. Studies by Wang and colleagues also showed that 90K inhibits HIV-1 virion production by interacting with Gag and vimentin (VIM) in trapping HIV-1 Gag to VIM filaments, suggesting an alternative anti-HIV-1 mechanism by 90K [46]. It is unknown whether HIV-1 has adopted any mechanisms to counter this factor. The antiviral function of 90K is conserved among primates except the rhesus macaque [47]. Interestingly, 90K impairs gp160 processing and reduces levels of gp120 on the plasma membrane in other species, including the rhesus macaque; however, these functions do not always reduce the infectivity of nascent HIV-1 virions [47]. Further studies have shown that the impairment of mature gp120 incorporation into virions might also contribute to the antiviral action of 90K [47].
IFITM3 also causes the accumulation of gp160 and loss of mature gp120/gp41 in HIV-1 particles [33]. IFITM3 is a member of the IFITM family, including IFITM1, IFITM2, IFITM3, IFITM5, and IFITM10. Among these, IFITM1, 2, and 3 are interferon-inducible and have been shown to inhibit a wide range of viruses (Reviewed in [48]). Ectopic expression of IFITM3, and to a lesser extent IFITM2, impairs gp160 cleavage, promotes gp120 shedding, and diminishes the level of mature gp120/gp41 in HIV-1 particles, thus reducing HIV-1 infectivity [33,49]. In departure from GBP5 and 90K, IFITM3 has been shown to associate with both gp160 and gp120/gp41, which may allow IFITM3 to directly interfere with processing of the gp160 precursor.
3.3. MARCH1, MARCH2, and MARCH8: Removing HIV-1 Env from the Cell Surface
Reaching the plasma membrane does not warrant safety for Env. MARCH8 (membrane-associated RING-CH 8) has been recently reported to modify HIV-1 Env and envelope proteins of other viruses to further downregulate them from the cell surface. MARCH8 is one of the 11 members of the MARCH family of RING-finger E3 ubiquitin ligases. As a transmembrane protein, MARCH8 bears a C4HC3 RING finger domain in the N-terminal cytoplasmic tail that recruits the E2 enzyme [50,51]. MARCH8 is involved in downregulating multiple transmembrane proteins, including but not limited to MHC-II [52], TRAIL receptor 1, and transferrin receptor [53]. Association of MARCH8 with these cellular transmembrane proteins often causes polyubiquitination of the target protein, followed by its trafficking to lysosomes for degradation. Another recently reported substrate for MARCH8 is BST-2, and MARCH8 regulates its ubiquitination, trafficking, and turnover [54]. Given its prolific regulation of cellular transmembrane proteins, it is thus not surprising that MARCH8 also targets viral envelope proteins and downregulates them from the cell surface [55].
When ectopically expressed, MARCH8 antagonizes not only HIV-1 Env but also glycoproteins of HIV-2, SIV, MLV, xenotropic MLV-related virus (XMRV), and VSV, suggesting a broad antiviral function. Mutating the RING domain, such as CS and W114A mutations, abrogates the antiviral activity of MARCH8, which demonstrates its dependence on E3 ligase activity. It is important to note that MARCH8 may impair different viral glycoproteins by different mechanisms. For example, MARCH8 removes HIV-1 Env from the cell surface, which is then retained within lysosomes without degradation. As a result, the total level of Env protein in cells does not change. In contrast, VSV G protein is downregulated by MARCH8 both at the cell surface and within the cell due to its degradation in lysosomes. Regardless of mechanistic details, MARCH8 interacts with both HIV-1 Env and VSV G proteins and likely alters their levels through ubiquitination. Further examination of antiviral activity exhibited by other members of the MARCH family revealed that MARCH1 and MARCH2 have antiviral functions similar to those observed in MARCH8; these members of MARCH family inhibit HIV-1 infectivity by downregulating HIV-1 Env from the cellular surface and reducing the levels of Env incorporated into the virions [56,57]. Similar to MARCH8, MARCH1 and MARCH2 are also localized at the plasma membrane [56]. As observed for MARCH8, MARCH1 also gets incorporated into virions; however, virion incorporation of MARCH2 remains controversial [56]. One group showed that HIV-1 infection increased MARHC2 expression but MARCH2 was not detected in the released virus particles [57], whereas another group showed that, similar to MARCH1 and MARCH8, MARCH2 is also found in progeny virions [56].
Higher levels of MARCH1, MARCH2, and MARCH8 were detected in myeloid cells such as monocyte-derived macrophages (MDM) and monocyte-derived dendritic cells (MDDCs) in comparison to primary CD4+ T cells. Unlike MARCH8, expression of MARCH1 and MARHC2 is highly inducible by type I IFN in MDM and MDDCs [55,56]. Knockdown or knockout of MARCH8 in myeloid cells increases HIV-1 infectivity, suggesting MARCH8 as one of the cellular factors that restrains HIV-1 infection of macrophages and dendritic cells. HIV-1 Vpr, Nef, and Vpu do not antagonize MARCH proteins; as a result, it remains to be determined how HIV-1 and other viruses, especially those that replicate in macrophages and dendritic cells, evade inhibition by MARCH1, MARCH2, and MARCH8. Since these MARCH proteins remove HIV-1 Env from the plasma membrane, it is speculated that HIV-1 takes advantage of these proteins to escape immunosurveillance.
4. Cellular Antagonists of Env Protein in HIV-1 Particles
In addition to targeting Env in the infected cells and preventing its incorporation into virus particles, some cellular factors such as IFITM3 and SERINC5 also find their way into virus particles and block the fusion of viral membrane and cellular membrane.
Beyond its impairment of gp160 processing in HIV-1 producing cells, IFITM3 is incorporated into HIV-1 particles. Virion incorporation of IFITM3 is at least partially due to its interaction with HIV-1 Env protein, as shown by co-immunoprecipitation [33,58]. Compared to IFITM3, IFITM2 demonstrates weaker inhibitory activity, whereas IFITM1 shows the least anti-HIV-1 activity [33,49,59]. One mechanism behind this impairment of HIV-1 infectivity is the reduction of gp120 in HIV-1 particles when HIV-1 is produced from 293T cells transfected with IFITM3 DNA and proviral DNA [33]. Alternative mechanisms may also exist, since IFITM3-bearing HIV-1 particles produced from CD4+ U87 cells are also less infectious but without detectable defects in viral Env protein [60]. In addition to HIV-1, IFITM proteins have also been detected in the particles of a large group of enveloped viruses, namely, murine leukemia virus (MLV), Mason–Pfizer monkey virus (MPMV), VSV, measles virus (MeV), Ebola virus (EBOV), West Nile virus (WNV), dengue virus (DENV), and Epstein–Barr virus (EBV), all leading to a decreased viral infectivity [61]. IFITM proteins’ broad spectrum of antiviral activity suggests a general mechanism that recruits IFITM proteins into different viruses to dampen viral infectivity.
Another factor in virions, SERINC5, is a member of the serine incorporator (SERINC) family. As its name indicates, SERINC proteins are involved in the synthesis of two serine-containing lipids: phosphatidylserine and sphingolipids [62]. In 2015, two groups reported that in the absence of HIV-1 Nef protein, SERINC5, and to a lesser extent SERINC3, is incorporated into HIV-1 particles and impairs HIV-1 infectivity [63,64]. SERINC5 contains 11 transmembrane domains, and is associated with lipid rafts where HIV-1 particles often form. Presence of SERINC5 in HIV-1 particles obstructs the formation of the viral fusion pore [65], thus inhibiting HIV-1 entry into target cells. This mechanism of action likely results from an increased rigidity of viral membrane that bears clustered SERINC5, rather than altered lipid composition of viral membrane by SERINC5 [66]. SERINC5 interferes with the conformation of the MPER region of Env, which may contribute to its inhibition of Env-mediated cellular entry [65,67]. HIV-1 and other viruses have evolved countermeasures to antagonize SERINC5. HIV-1 Nef protein downregulates SERINC5 from the plasma membrane via the endosome/lysosome pathway, thus preventing SERINC5 incorporation into HIV-1 particles [68]. Interestingly, the Env protein of some HIV-1 strains are refractory to SERINC5 inhibition, which is analogous to the SERINC5-resistant property of VSV G, Ebola GP, and other viral envelope proteins [64]. In addition to these viral antagonists, the glycoGag protein of gammaretroviruses (such as MLV) and the S2 protein of equine infectious anemia virus (EIAV) have also been reported to overcome the antiviral function of SERINC5 [69,70]. The anti-SERINC5 strategies from different viruses indicate its broad antiviral function.
5. Env Antagonists in the Membrane of Target Cells: The Other Half of the Fusion Story
HIV-1 entry is marked by the fusion of viral and target cell membranes; this process is driven by the sequential conformational changes of Env trimer as a result of binding to receptor CD4 and co-receptor CCR5 or CXCR4. In addition to the series of host inhibitory mechanisms discussed above present in virus producing cells that target and disable Env in the viral membrane, target cell membrane is also equipped with mechanisms to prevent fusion with viral membrane. One prominent example is the antiviral function of IFITM proteins, which was discovered in a genome-wide siRNA screen for host factors that modulate the infection of influenza A virus [71]. Subsequently, IFITM proteins were shown to inhibit HIV-1 entry in a shRNA-based screen aiming to identify anti-HIV-1 ISGs [14]. Mechanistic studies further revealed that these IFITM proteins hamper viral membrane hemi-fusion and/or block the formation of fusion pore in virus target cells, due to the increased rigidity and altered curvature of IFITM-bearing cellular membranes [72,73]. IFITM proteins may modulate membrane fluidity by interfering with intracellular cholesterol homeostasis [72,73,74,75], although the involvement of cholesterol is still controversial [75]. Entry deterrence of incoming viruses also benefits from the subcellular localization of IFITM proteins at the plasma membrane and in endosomes/lysosomes, which covers the route of virus entry [76,77,78]. The N-terminal sequences of IFITM2 and IFITM3 bear the YMEL motif that binds to AP-2, and guides its endosomal and lysosomal localization via the endocytic pathway [78]. In contrast, IFITM1 lacks this endocytic motif; rather, it carries a KR-sorting motif at the C-terminus, which guides IFITM1 trafficking to recycling/early endosomes [79,80].
IFITM proteins are not the sole defense in target cells against virus entry. Cholesterol-25-Hydroxylase (CH25H), another ISG, protects target cells from viral infection [81]. CH25H produces a soluble oxysterol, 25-hydroxycholesterol (25-HC), which inhibits a large group of viruses including murine herpesvirus 68 (MHV68), VSV, Zika virus, HIV-1, herpes simplex virus 1 (HSV-1), EBOV, Nipah virus, Russian Spring–summer encephalitis virus, Rift Valley fever virus, and hepatitis C virus [82,83,84,85]. In addition to protecting its producer cells, 25-HC can be secreted to restrict virus entry into surrounding uninfected cells [83]. While 25-HC has been reported to regulate cholesterol biosynthesis and maintain cholesterol homeostasis, this function has been challenged by the observation that CH25H-deficient mice demonstrated normal cholesterol metabolism [86,87,88]. Clinical evidence from patients suffering from a hereditary disease, spastic paresis, that displays a high level of 25-HC but a normal level of cholesterol further disputes the role of 25-HC in cholesterol regulation. In contrast, accumulating evidence suggests an upregulation of CH25H in macrophages and dendritic cells upon inflammatory stimulation [81,89,90]. Furthermore, it was reported that accumulation of 25-HC instead of cholesterol in the lipid membrane prevents HIV-1 Env-mediated membrane fusion by modifying the secondary structure of the HIV-fusion peptide [91]. Analogous to IFITM proteins, 25-HC also operates in virus-producing cells by altering the glycosylation of Lassa virus glycoprotein [92]. Interestingly, 25-HC does not spare non-enveloped viruses, unlike IFITM proteins [93]. For example, 25-HC was shown to hamper reovirus uncoating [93]. Again, IFITM3 and 25-HC are thematically resonant in their antiviral actions, given the observation that IFITM3 likely prevents reovirus entry by delaying the proteolytic processing of reovirus particles within late endosomes [94]. Independent to producing 25-HC, CH25H also operates by directly acting on viral proteins. For example, the catalytically inactive CH25H mutant retains its antiviral function against HCV and porcine reproductive and respiratory syndrome virus (PRRSV) through direct interactions with NS5A of HCV and nsp1α of PRRSV [84].
6. Env Protein Fights Back: Evasion of Host Restriction on Virus Entry and Beyond
In order to replicate and transmit, viruses need to counter and evade the multi-layered host restriction defense system. Identification of viral antagonism against a host restriction factor also demonstrates the presence of this restriction in the context of in vivo viral infections, which have driven the selection and evolution of specific viral counter measures. Indeed, viral antagonistic strategies have been discovered for some host restriction mechanisms targeting HIV-1 entry, which began to illuminate the diversity of viral evolution in evading host restriction.
One viral countermeasure is to use a viral protein to target and downregulate the host restriction factor, which is well illustrated by the downregulation of SERINC5 by HIV-1 Nef, MLV glycoGag, and EIAV S2 proteins. A second strategy is to counter restriction factor inhibition through an indirect escape mechanism. One example of this mechanism is to increase the level of Env expression to counter host inhibition of virus entry, exemplified by HIV-1 escape from GBP5 inhibition through shutting down Vpu to elevate Env expression. Another example of viral adaptation and escape has been shown through Vif-null HIV-1 viruses conferring full resistance to APOBEC3G (A3G), which has been linked to a novel Env-dependent mechanism [95]. Env adaptation in Vif-null HIV-1 virus decreases virus fusogenicity and leads to higher levels of Gag-pol packaging into virions, which increases the levels of reverse transcriptase (RT). This Env-mediated elevation in RT levels prevents A3G-mediated hypermutation [95].
The third known strategy is to change Env protein sequence and thus adjust its entry function to gain resistance to host restriction of virus entry. We observed this viral escape mechanism when passaging HIV-1 in IFITM1-expressing SupT1 cells with the goal to select IFITM1-resistant viruses. The resistance mutations that enhanced HIV-1 cell-to-cell transmission were identified in viral Vpu and Env proteins, and they rescued HIV-1 replication in IFITM1-expression SupT1 cells [96]. Similarly, we identified Env mutations that enhance HIV-1 replication in IFITM3-expresing cells [33]. We later found that changing the V3 loop alone in Env can confer resistance to IFITM3 inhibition [49]. The ability of HIV-1 Env protein to resist IFITM3 was also observed in the transmitted founder HIV-1 strains [60]. This study convincingly showed that Env mutations, which arise to evade autologous antibodies 6 months after infection, transform the IFITM3-resistant HIV-1 to an IFITM3-sensitive one. Molecular and structural features of HIV-1 Env that determine its susceptibility and resistance to IFITM3 inhibition remain to be fully elucidated. A related envelope protein-mediated evasion was reported for influenza A virus [97]. It is known that IAV tends to finalize membrane fusion at a low pH in late endosomes/lysosomes, where IFITM3 is abundantly present. To escape from IFITM3 restriction, IAV HA protein can adapt to mediate membrane fusion in early endosomes where pH is relatively less acidic and has low levels of IFITM3 [97].
It appears that viral envelope protein resists more than just IFITM proteins. We and others have found that Env proteins of some HIV-1 strains, including transmitted founder HIV-1 isolates, are able to resist SERINC5 inhibition even in the absence of viral Nef protein which acts as a SERINC5 antagonist [64,67]. We further mapped the resistant determinant to the V3 loop of Env. Viral envelope has also demonstrated the capacity of overcoming host restrictions beyond virus entry. For example, passage of SIV/HIV chimeric virus (SHIV) in macaques in the presence of interferon-α led to the selection of interferon-α-resistant SHIV [98]. This resistance phenotype was mapped to viral Env protein that had a higher level of expression from the resistant virus. A separate study reported resistance of transmitted founder HIV-1 to type II interferon (interferon-γ), and this resistance activity was also mapped to viral Env [99]. In support of Env’s role in countering interferon-γ, replication of the sensitive HIV-1 strain in the presence of interferon-γ selected for resistance mutations in viral Env protein [99]. These studies report a general role of HIV-1 Env protein in generating resistance to interferon suppression. One possibility is that HIV-1 changes Env to acquire higher replication capacity in compensating for the loss of infectivity as a result of interferon inhibition.
In addition to this compensatory mechanism, viral envelope proteins are able to directly counter specific host restriction factors. One example is the antagonization of tetherin by HIV-2 Env, HERV-K Env, and Ebola glycoprotein [100,101,102]. Tetherin is known to inhibit the release of HIV-1 and many other enveloped viruses by tethering the progeny virions to the cell surface [103,104]. HIV-1 uses Vpu to nullify tetherin, while some primate lentiviruses such as SIVs from chimpanzee, sooty mangabeys, and African green monkeys use Nef to antagonize tetherin [105]. In contrast, HIV-2 and some SIV lineages including SIV tetanus do not encode for Vpu, they instead use Env as a tetherin antagonist [100,106]. Functional domain analysis revealed that Env’s antagonist activity against tetherin depends on a tyrosine based motif (YXX) in the cytoplasmic tail of the gp41 membrane proximal region [100]. Upon direct interaction between the extracellular domain of Env and the ectodomain of tetherin, the tyrosine motif (YXX of Env gp41 recruits AP-2 complex and induces intracellular sequestration of tetherin from the cell surface and its accumulation in the trans-Golgi network [100,107,108]. Recent studies on HIV-2 isolates from different patients reveal that the anti-tetherin activity is a conserved function of HIV-2 Env [109]. Some ancient retroviruses might have also used their Env proteins to overcome tetherin inhibition, since the youngest and most active endogenous retrovirus (ERV) in human genomes, HERV-K, still preserves this function through its Env protein [101]. In addition to retroviruses, the glycoproteins (GPs) of Ebola virus and Lluvia virus are also antagonists of tetherin. Ebola GP does not remove tetherin from the cell surface but appears to depend on a GxxxA motif in its transmembrane domain [110].
7. Viral Envelope Protein under the Suppressive Pressure of Both Adaptive and Innate Immunity
Adaptive and innate immunity cooperate to create a higher genetic barrier for viral envelope protein to escape compared to either immune response alone. For example, HIV-1 Env is engaged in a constant battle with the antibody-mediated adaptive immune response. The ability of Env to evade an innate immunity might be limited by the need to resist antibody attack. This scenario is illustrated by the loss of IFITM3 resistance in transmitted founder HIV-1 strains, which mutate Env in order to resist autologous antibodies as infection progresses [60]. One implication of this finding is that relatively high level of IFITM3 may mount high enough inhibitory pressure to limit Env mutation pathways in evading inhibition by antibodies, thus creating a synergistic control of HIV-1 infection. It is equally possible that given the interferon-inducible nature of IFITM3 expression, subsidence of interferon response after the acute stage of HIV-1 infection may lead to reduction in IFITM3 level, thus mounting less pressure on HIV-1 Env and allowing Env to change and resist neutralizing antibodies.
The potential interplay of adaptive and innate immunity may also explain the need for Nef to antagonize SERINC5, even though HIV-1 Env has full capacity of SERINC5 resistance. This requirement is likely because certain types of neutralizing antibodies, such as those targeting the MPER sequence of Env, are able to sensitize the otherwise resistant Env to SERINC5 inhibition. This example demonstrates that dual pressures from SERINC5 (innate immunity) and neutralizing antibodies (adaptive immunity) have driven HIV-1 to evolve Nef’s antagonism against SERINC5. Further research could illuminate the synergistic suppression on viral envelope protein from both adaptive and innate immunity, and the seemingly endless evolution of viral counter measures to ensure viral survival.
8. Conclusions
A growing body of studies demonstrate that HIV-1 Env protein is not only the primary antigen of adaptive immunity but also the main target of innate immunity. An arsenal of antiviral proteins have already been discovered that either limit the synthesis of Env protein, deregulate Env glycosylation, impair Env cleavage by furin, or impede the incorporation of mature Env trimers into HIV-1 particles. In addition, some antiviral factors, such as SERINC5, IFITM3, and 25-HC restrict HIV-1 entry not by acting on Env directly but by altering the physical property of viral membranes or cellular membranes, which often enable them to inhibit a broad range of viruses. Cells use these diverse molecular mechanisms to inhibit the entry of many viruses far beyond HIV-1, which illustrates that targeting virus entry is a general and important host antiviral strategy (Figure 1, Table 1). Future research is expected to provide further insights into the molecular mechanisms by which each of these antiviral proteins restricts virus entry, to elucidate how these factors function together in vivo to create an optimal antiviral effect, and to understand viral countermeasures and escape mechanisms. It will also be interesting to investigate how these antiviral proteins, through altering the Env glycoprotein, modulate adaptive immune responses. The bnAbs, key effectors in adaptive immunity, are now being tested in clinical trials as a new HIV treatment [111]. Likewise, restriction factors, forming a key layer of innate immunity, also promise new approaches to treat and even cure HIV infection, such as the application of TRIM5α in gene therapy to create HIV-resistant hematopoietic stem cells [112,113].
Author Contributions
S.B., Y.W., S.-L.L., and C.L. prepared the manuscript.
Acknowledgments
This work was supported by funding from Canadian Institutes of Health Research (MOP-133479) to C.L. The work of S.-L.L. was supported by NIH was supported by NIH grants R01AI112381 and R01 GM132069. We are grateful to Judy Huang and Victoria Leopardi for critical reading and editing of the manuscript.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 Envelope Glycoprotein Biosynthesis, Trafficking, and Incorporation. J. Mol. Boil. 2011, 410, 582–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiñones-Kochs, M.I.; Buonocore, L.; Rose, J.K. Role of N-Linked Glycans in a Human Immunodeficiency Virus Envelope Glycoprotein: Effects on Protein Function and the Neutralizing Antibody Response. J. Virol. 2002, 76, 4199–4211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.R.; Mascola, J.R. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat. Immunol. 2015, 16, 571–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardine, J.G.; Ota, T.; Sok, D.; Pauthner, M.; Kulp, D.W.; Kalyuzhniy, O.; Skog, P.D.; Thinnes, T.C.; Bhullar, D.; Briney, B.; et al. HIV-1 VACCINES. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 2015, 349, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.; Rollman, E.; Johansson, S.; Kent, S.; Stratov, I. The Utility of ADCC Responses in HIV Infection. Curr. HIV Res. 2008, 6, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; De Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Prévost, J.; Alsahafi, N.; Ding, S.; Finzi, A. Impact of HIV-1 Envelope Conformation on ADCC Responses. Trends Microbiol. 2018, 26, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Sparrer, K.M.; Gack, M.U. Intracellular detection of viral nucleic acids. Curr. Opin. Microbiol. 2015, 26, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef]
- Degols, G.; Eldin, P.; Mechti, N. ISG20, an actor of the innate immune response. Biochimie 2007, 89, 831–835. [Google Scholar] [CrossRef]
- Refsland, E.W.; Harris, R.S. The APOBEC3 Family of Retroelement Restriction Factors. Curr. Top. Microbiol. Immunol. 2013, 371, 1–27. [Google Scholar] [Green Version]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. Erratum: SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef]
- MacMicking, J.D. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 2012, 12, 367–382. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.-L.; Liang, C. The IFITM Proteins Inhibit HIV-1 Infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012, 491, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Clerzius, G.; Gelinas, J.F.; Gatignol, A. Multiple levels of PKR inhibition during HIV-1 replication. Rev. Med. Virol. 2011, 21, 42–53. [Google Scholar] [CrossRef]
- Qi, M.; Williams, J.A.; Chu, H.; Chen, X.; Wang, J.-J.; Ding, L.; Akhirome, E.; Wen, X.; Lapierre, L.A.; Goldenring, J.R.; et al. Rab11-FIP1C and Rab14 Direct Plasma Membrane Sorting and Particle Incorporation of the HIV-1 Envelope Glycoprotein Complex. PLoS Pathog. 2013, 9, e1003278. [Google Scholar] [CrossRef]
- Kirschman, J.; Qi, M.; Ding, L.; Hammonds, J.; Dienger-Stambaugh, K.; Wang, J.-J.; Lapierre, L.A.; Goldenring, J.R.; Spearman, P. HIV-1 Envelope Glycoprotein Trafficking through the Endosomal Recycling Compartment Is Required for Particle Incorporation. J. Virol. 2018, 92, e01893-17. [Google Scholar] [CrossRef]
- Kluge, S.F.; Sauter, D.; Kirchhoff, F. SnapShot: Antiviral Restriction Factors. Cell 2015, 163, 774. [Google Scholar] [CrossRef]
- Land, A.; Zonneveld, D.; Braakman, I. Folding of HIV-1 Envelope glycoprotein involves extensive isomerization of disulfide bonds and conformation-dependent leader peptide cleavage. FASEB J. 2003, 17, 1058–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Davies, G. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Boil. 1997, 7, 637–644. [Google Scholar] [CrossRef]
- Gonzalez, D.S.; Karaveg, K.; Vandersall-Nairn, A.S.; Lal, A.; Moremen, K.W. Identification, Expression, and Characterization of a cDNA Encoding Human Endoplasmic Reticulum Mannosidase I, the Enzyme That Catalyzes the First Mannose Trimming Step in Mammalian Asn-linked Oligosaccharide Biosynthesis. J. Boil. Chem. 1999, 274, 21375–21386. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Frabutt, D.A.; Moremen, K.W.; Zheng, Y.H. ER ManI (Endoplasmic Reticulum Class I alpha-Mannosidase) Is Required for HIV-1 Envelope Glycoprotein Degradation via Endoplasmic Reticulum-associated Protein Degradation Pathway. J. Biol. Chem. 2015, 290, 22184–22192. [Google Scholar] [CrossRef]
- Zhou, T.; Dang, Y.; Zheng, Y.-H.; Sundquist, W.I. The Mitochondrial Translocator Protein, TSPO, Inhibits HIV-1 Envelope Glycoprotein Biosynthesis via the Endoplasmic Reticulum-Associated Protein Degradation Pathway. J. Virol. 2014, 88, 3474–3484. [Google Scholar] [CrossRef] [Green Version]
- Frabutt, D.A.; Wang, B.; Riaz, S.; Schwartz, R.C.; Zheng, Y.H. Innate Sensing of Influenza A Virus Hemagglutinin Glycoproteins by the Host Endoplasmic Reticulum (ER) Stress Pathway Triggers a Potent Antiviral Response via ER-Associated Protein Degradation. J. Virol. 2018, 92, e01690-17. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, T.; Frabutt, D.A.; Zheng, Y.-H. HIV-1 Vpr increases Env expression by preventing Env from endoplasmic reticulum-associated protein degradation (ERAD). Virology 2016, 496, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Casini, A.; Olivieri, M.; Vecchi, L.; Burrone, O.R.; Cereseto, A. Reduction of HIV-1 infectivity through endoplasmic reticulum-associated degradation-mediated Env depletion. J. Virol. 2015, 89, 2966–2971. [Google Scholar] [CrossRef] [PubMed]
- Jejcic, A.; Daniels, R.; Goobar-Larsson, L.; Hebert, D.N.; Vahlne, A. Small Molecule Targets Env for Endoplasmic Reticulum-Associated Protein Degradation and Inhibits Human Immunodeficiency Virus Type 1 Propagation. J. Virol. 2009, 83, 10075–10084. [Google Scholar] [CrossRef] [PubMed]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human WD protein, h-βTrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Krapp, C.; Hotter, D.; Gawanbacht, A.; McLaren, P.J.; Kluge, S.F.; Stürzel, C.M.; Mack, K.; Reith, E.; Engelhart, S.; Ciuffi, A.; et al. Guanylate Binding Protein (GBP) 5 Is an Interferon-Inducible Inhibitor of HIV-1 Infectivity. Cell Host Microbe 2016, 19, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Lodermeyer, V.; Suhr, K.; Schrott, N.; Kolbe, C.; Stürzel, C.M.; Krnavek, D.; Münch, J.; Dietz, C.; Waldmann, T.; Kirchhoff, F.; et al. 90K, an interferon-stimulated gene product, reduces the infectivity of HIV-1. Retrovirology 2013, 10, 111. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.-M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. IFITM Proteins Restrict HIV-1 Infection by Antagonizing the Envelope Glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Vestal, D.J.; Jeyaratnam, J.A. The Guanylate-Binding Proteins: Emerging Insights into the Biochemical Properties and Functions of This Family of Large Interferon-Induced Guanosine Triphosphatase. J. Interface Cytokine Res. 2011, 31, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.H.; Shenoy, A.R.; Kumar, P.; Bradfield, C.J.; MacMicking, J.D. IFN-inducible GTPases in host cell defense. Cell Host Microbe 2012, 12, 432–444. [Google Scholar] [CrossRef]
- Pan, W.; Zuo, X.; Feng, T.; Shi, X.; Dai, J. Guanylate-binding protein 1 participates in cellular antiviral response to dengue virus. Virol. J. 2012, 9, 292. [Google Scholar] [CrossRef]
- Anderson, S.L.; Carton, J.M.; Lou, J.; Xing, L.; Rubin, B.Y. Interferon-Induced Guanylate Binding Protein-1 (GBP-1) Mediates an Antiviral Effect against Vesicular Stomatitis Virus and Encephalomyocarditis Virus. Virology 1999, 256, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, P.J.; Gawanbacht, A.; Pyndiah, N.; Krapp, C.; Hotter, D.; Kluge, S.F.; Götz, N.; Heilmann, J.; Mack, K.; Sauter, D.; et al. Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology 2015, 12, 41. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Hizukuri, Y.; Yamashiro, K.; Makita, N.; Ohnishi, K.; Takeya, M.; Komohara, Y.; Hayashi, Y. Guanylate-binding protein 5 is a marker of interferon-gamma-induced classically activated macrophages. Clin. Transl. Immunol. 2016, 5, e111. [Google Scholar] [CrossRef]
- Guerrero, S.; Batisse, J.; Libre, C.; Bernacchi, S.; Marquet, R.; Paillart, J.-C.; Boehr, D. HIV-1 Replication and the Cellular Eukaryotic Translation Apparatus. Viruses 2015, 7, 199–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, E.R.; Dunfee, R.L.; Stanton, J.; Bogdan, D.; Kunstman, K.; Wolinsky, S.M.; Gabuzda, D. High Frequency of Defective vpu Compared with tat and rev Genes in Brain from Patients with HIV Type 1-Associated Dementia. AIDS Res. Hum. Retrovir. 2007, 23, 575–580. [Google Scholar] [CrossRef]
- Brakebusch, C.; Sures, I.; Jallal, B.; Mossie, K.; Fusco, O.; Iacobelli, S.; Ullrich, A. Isolation and Functional Characterization of the Human 90K Promoter. Genomics 1999, 57, 268–278. [Google Scholar] [CrossRef]
- Briggs, N.C.; Natoli, C.; Tinari, N.; D’egidio, M.A.; Goedert, J.J.; Iacobelli, S. A 90-kDa protein serum marker for the prediction of progression to AIDS in a cohort of HIV-1+ homosexual men. AIDS Res. Hum. Retrovir. 1993, 9, 811–816. [Google Scholar] [CrossRef]
- Darcissac, E.C.A.; Vidal, V.; De La Tribonnière, X.; Mouton, Y.; Bahr, G.M. Variations in serum IL-7 and 90K/Mac-2 binding protein (Mac−2 BP) levels analysed in cohorts of HIV-1 patients and correlated with clinical changes following antiretroviral therapy. Clin. Exp. Immunol. 2001, 126, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Loh, L.C.; Keeler, V.; Laferte, S. Monoclonal antibodies specific for human tumor-associated antigen 90K/Mac-2 binding protein: Tools to examine protein conformation and function. J. Cell. Biochem. 2000, 77, 540–559. [Google Scholar]
- Wang, Q.; Zhang, X.; Han, Y.; Wang, X.; Gao, G. M2BP inhibits HIV-1 virion production in a vimentin filaments-dependent manner. Sci. Rep. 2016, 6, 32736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodermeyer, V.; Ssebyatika, G.; Passos, V.; Ponnurangam, A.; Malassa, A.; Ewald, E.; Stürzel, C.M.; Kirchhoff, F.; Rotger, M.; Falk, C.S.; et al. The Antiviral Activity of the Cellular Glycoprotein LGALS3BP/90K Is Species Specific. J. Virol. 2018, 92, e00226-18. [Google Scholar] [CrossRef]
- Bailey, C.C.; Zhong, G.; Huang, I.-C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Pan, Q.; Ding, S.; Wang, Z.; Yu, J.; Finzi, A.; Liu, S.-L.; Liang, C. The V3 Loop of HIV-1 Env Determines Viral Susceptibility to IFITM3 Impairment of Viral Infectivity. J. Virol. 2017, 91, e02441-16. [Google Scholar] [CrossRef]
- Bartee, E.; Mansouri, M.; Nerenberg, B.T.H.; Gouveia, K.; Früh, K. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol. 2004, 78, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Goto, E.; Ishido, S.; Sato, Y.; Ohgimoto, S.; Ohgimoto, K.; Nagano-Fujii, M.; Hotta, H. c-MIR, a Human E3 Ubiquitin Ligase, Is a Functional Homolog of Herpesvirus Proteins MIR1 and MIR2 and Has Similar Activity. J. Boil. Chem. 2003, 278, 14657–14668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Shin, J.-S. Molecular mechanism and cellular function of MHCII ubiquitination. Immunol. Rev. 2015, 266, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Iwabu, Y.; Tokunaga, K.; Tanaka, Y. Membrane-associated RING-CH (MARCH) 8 mediates the ubiquitination and lysosomal degradation of the transferrin receptor. J. Cell Sci. 2013, 126, 2798–2809. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.; Pacini, G.; Berlioz-Torrent, C.; Janvier, K. Characterization of E3 ligases involved in lysosomal sorting of the HIV-1 restriction factor BST2. J. Cell Sci. 2017, 130, 1596–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, T.; Zhang, Y.; Koyama, T.; Tobiume, M.; Tsunetsugu-Yokota, Y.; Yamaoka, S.; Fujita, H.; Tokunaga, K. MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat. Med. 2015, 21, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tada, T.; Ozono, S.; Yao, W.; Tanaka, M.; Yamaoka, S.; Kishigami, S.; Fujita, H.; Tokunaga, K. Membrane-associated RING-CH (MARCH) 1 and 2 are MARCH family members that inhibit HIV-1 infection. J. Boil. Chem. 2019, 294, 3397–3405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, J.; Liu, X. MARCH2 is upregulated in HIV-1 infection and inhibits HIV-1 production through envelope protein translocation or degradation. Virology 2018, 518, 293–300. [Google Scholar] [CrossRef]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.-N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 2014, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM Proteins Incorporated into HIV-1 Virions Impair Viral Fusion and Spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of Transmitted Founder HIV-1 to IFITM-Mediated Restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Tartour, K.; Nguyen, X.-N.; Appourchaux, R.; Assil, S.; Barateau, V.; Bloyet, L.-M.; Gaillard, J.B.; Confort, M.-P.; Escudero-Perez, B.; Gruffat, H.; et al. Interference with the production of infectious viral particles and bimodal inhibition of replication are broadly conserved antiviral properties of IFITMs. PLoS Pathog. 2017, 13, e1006610. [Google Scholar] [CrossRef]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an Activity-regulated Protein Family, Incorporates Serine into Membrane Lipid Synthesis. J. Boil. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Boil. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef] [Green Version]
- Trautz, B.; Wiedemann, H.; Lüchtenborg, C.; Pierini, V.; Kranich, J.; Glass, B.; Kräusslich, H.-G.; Brocker, T.; Pizzato, M.; Ruggieri, A.; et al. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J. Boil. Chem. 2017, 292, 13702–13713. [Google Scholar] [CrossRef] [PubMed]
- Beitari, S.; Ding, S.; Pan, Q.; Finzi, A.; Liang, C. Effect of HIV-1 Env on SERINC5 Antagonism. J. Virol. 2017, 91, e02214-16. [Google Scholar] [CrossRef]
- Shi, J.; Xiong, R.; Zhou, T.; Su, P.; Zhang, X.; Qiu, X.; Li, H.; Li, S.; Yu, C.; Wang, B.; et al. HIV-1 Nef Antagonizes SERINC5 Restriction by Downregulation of SERINC5 via the Endosome/Lysosome System. J. Virol. 2018, 92, e00196-18. [Google Scholar] [CrossRef]
- Ahi, Y.S.; Zhang, S.; Thappeta, Y.; Denman, A.; Feizpour, A.; Gummuluru, S.; Reinhard, B.; Muriaux, D.; Fivash, M.J.; Rein, A. Functional Interplay Between Murine Leukemia Virus Glycogag, Serinc5, and Surface Glycoprotein Governs Virus Entry, with Opposite Effects on Gammaretroviral and Ebolavirus Glycoproteins. mBio 2016, 7, e01985-16. [Google Scholar] [CrossRef] [PubMed]
- Chande, A.; Cuccurullo, E.C.; Rosa, A.; Ziglio, S.; Carpenter, S.; Pizzato, M. S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors SERINC5 and SERINC3. Proc. Natl. Acad. Sci. USA 2016, 113, 13197–13202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; Van Der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef]
- Li, K.; Markosyan, R.M.; Zheng, Y.-M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM Proteins Restrict Viral Membrane Hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 Restricts Influenza A Virus Entry by Blocking the Formation of Fusion Pores following Virus-Endosome Hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef] [PubMed]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.-C.; Farzan, M.; Jung, J.U. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrensch, F.; Winkler, M.; Pöhlmann, S. IFITM Proteins Inhibit Entry Driven by the MERS-Coronavirus Spike Protein: Evidence for Cholesterol-Independent Mechanisms. Viruses 2014, 6, 3683–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, A.R.; Reid, L.E.; McMahon, M.; Stark, G.R.; Kerr, I.M. Molecular analysis of a human interferon-inducible gene family. Eur. J. Biochem. 1991, 199, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, R.; Pan, Q.; Ding, S.; Rong, L.; Liu, S.-L.; Geng, Y.; Qiao, W.; Liang, C. The N-Terminal Region of IFITM3 Modulates Its Antiviral Activity by Regulating IFITM3 Cellular Localization. J. Virol. 2012, 86, 13697–13707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, R.; Xu, F.; Qian, J.; Yao, Y.; Miao, C.; Zheng, Y.-M.; Liu, S.-L.; Guo, F.; Geng, Y.; Qiao, W.; et al. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell. Microbiol. 2014, 16, 1080–1093. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.C.; Kondur, H.R.; Huang, I.-C.; Farzan, M. Interferon-induced Transmembrane Protein 3 Is a Type II Transmembrane Protein. J. Boil. Chem. 2013, 288, 32184–32193. [Google Scholar] [CrossRef]
- Li, K.; Jia, R.; Li, M.; Zheng, Y.M.; Miao, C.; Yao, Y.; Ji, H.L.; Geng, Y.; Qiao, W.; Albritton, L.M.; et al. A sorting signal suppresses IFITM1 restriction of viral entry. J. Biol. Chem. 2015, 290, 4248–4259. [Google Scholar] [CrossRef]
- Park, K.; Scott, A.L. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J. Leukoc. Boil. 2010, 88, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Lund, E.G.; Kerr, T.A.; Sakai, J.; Li, W.P.; Russell, D.W. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J. Biol. Chem. 1998, 273, 34316–34327. [Google Scholar] [CrossRef]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, S.; Yi, Z.; Tian, H.; Aliyari, R.; Li, Y.; Chen, G.; Liu, P.; Zhong, J.; Chen, X.; et al. Interferon-Inducible Cholesterol-25-Hydroxylase Inhibits Hepatitis C Virus Replication via Distinct Mechanisms. Sci. Rep. 2014, 4, 7242. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Deng, Y.-Q.; Wang, S.; Ma, F.; Aliyari, R.; Huang, X.-Y.; Zhang, N.-N.; Watanabe, M.; Dong, H.-L.; Liu, P.; et al. 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity 2017, 46, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Kandutsch, A.A.; Chen, H.W. Inhibition of sterol synthesis in cultured mouse cells by cholesterol derivatives oxygenated in the side chain. J. Boil. Chem. 1974, 249, 6057–6061. [Google Scholar]
- Goldstein, J.L.; Brunschede, G.Y.; Brown, M.S. Inhibition of proteolytic degradation of low density lipoprotein in human fibroblasts by chloroquine, concanavalin A, and Triton WR 1339. J. Boil. Chem. 1975, 250, 7854–7862. [Google Scholar]
- McDonald, J.G.; Russell, D.W. Editorial: 25-Hydroxycholesterol: A new life in immunology. J. Leukoc. Boil. 2010, 88, 1071–1072. [Google Scholar] [CrossRef]
- Diczfalusy, U.; Olofsson, K.E.; Carlsson, A.-M.; Gong, M.; Golenbock, D.T.; Rooyackers, O.; Fläring, U.; Björkbacka, H. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J. Res. 2009, 50, 2258–2264. [Google Scholar] [CrossRef] [Green Version]
- Bauman, D.R.; Bitmansour, A.D.; McDonald, J.G.; Thompson, B.M.; Liang, G.; Russell, D.W. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. USA 2009, 106, 16764–16769. [Google Scholar] [CrossRef] [Green Version]
- Gomes, B.; Gonçalves, S.; Disalvo, A.; Hollmann, A.; Santos, N.C. Effect of 25-hydroxycholesterol in viral membrane fusion: Insights on HIV inhibition. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1171–1178. [Google Scholar] [CrossRef]
- Shrivastava-Ranjan, P.; Bergeron, É.; Chakrabarti, A.K.; Albariño, C.G.; Flint, M.; Nichol, S.T.; Spiropoulou, C.F. 25-Hydroxycholesterol Inhibition of Lassa Virus Infection through Aberrant GP1 Glycosylation. mBio 2016, 7, e01808-16. [Google Scholar] [CrossRef]
- Doms, A.; Sanabria, T.; Hansen, J.N.; Altan-Bonnet, N.; Holm, G.H. 25-Hydroxycholesterol Production by the Cholesterol-25-Hydroxylase Interferon-Stimulated Gene Restricts Mammalian Reovirus Infection. J. Virol. 2018, 92, e01047-18. [Google Scholar] [CrossRef]
- Anafu, A.A.; Bowen, C.H.; Chin, C.R.; Brass, A.L.; Holm, G.H. Interferon-inducible Transmembrane Protein 3 (IFITM3) Restricts Reovirus Cell Entry. J. Boil. Chem. 2013, 288, 17261–17271. [Google Scholar] [CrossRef]
- Ikeda, T.; Symeonides, M.; Albin, J.S.; Li, M.; Thali, M.; Harris, R.S. HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PLoS Pathog. 2018, 14, e1007010. [Google Scholar] [CrossRef]
- Ding, S.; Pan, Q.; Liu, S.-L.; Liang, C. HIV-1 mutates to evade IFITM1 restriction. Virology 2014, 454, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, T.; Hensen, L.; Matrosovich, T.; Bergmann, J.; Winkler, M.; Peteranderl, C.; Klenk, H.-D.; Weber, F.; Herold, S.; Pöhlmann, S.; et al. pH Optimum of Hemagglutinin-Mediated Membrane Fusion Determines Sensitivity of Influenza A Viruses to the Interferon-Induced Antiviral State and IFITMs. J. Virol. 2017, 91, e00246-17. [Google Scholar] [CrossRef]
- Boyd, D.F.; Sharma, A.; Humes, D.; Cheng-Mayer, C.; Overbaugh, J. Adapting SHIVs In Vivo Selects for Envelope-Mediated Interferon-alpha Resistance. PLoS Pathog. 2016, 12, e1005727. [Google Scholar] [CrossRef]
- Rihn, S.J.; Foster, T.L.; Busnadiego, I.; Aziz, M.A.; Hughes, J.; Neil, S.J.D.; Wilson, S.J. The Envelope Gene of Transmitted HIV-1 Resists a Late Interferon Gamma-Induced Block. J. Virol. 2017, 91, e02254-16. [Google Scholar] [CrossRef]
- Le Tortorec, A.; Neil, S.J.D. Antagonism to and Intracellular Sequestration of Human Tetherin by the Human Immunodeficiency Virus Type 2 Envelope Glycoprotein. J. Virol. 2009, 83, 11966–11978. [Google Scholar] [CrossRef]
- Lemaître, C.; Harper, F.; Pierron, G.; Heidmann, T.; Dewannieux, M.; Ross, S.R.; Palmer, S.G.; DeVito, I.; Jenkins, S.G.; Niewiesk, S.; et al. The HERV-K Human Endogenous Retrovirus Envelope Protein Antagonizes Tetherin Antiviral Activity. J. Virol. 2014, 88, 13626–13637. [Google Scholar] [CrossRef] [Green Version]
- Lopez, L.A.; Yang, S.J.; Hauser, H.; Exline, C.M.; Haworth, K.G.; Oldenburg, J.; Cannon, P.M. Ebola Virus Glycoprotein Counteracts BST-2/Tetherin Restriction in a Sequence-Independent Manner That Does Not Require Tetherin Surface Removal. J. Virol. 2010, 84, 7243–7255. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wilson, S.J.; Langford, W.; Virgen, B.; Gregory, D.; Johnson, M.C.; Münch, J.; Kirchhoff, F.; Bieniasz, P.D.; Hatziioannou, T.; et al. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 2009, 6, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Mlcochova, P.; Pelchen-Matthews, A.; Petit, S.J.; Mattiuzzo, G.; Pillay, D.; Takeuchi, Y.; Marsh, M.; Towers, G.J.; Towers, G. Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST-2/CD317 by intracellular sequestration. Proc. Natl. Acad. Sci. USA 2009, 106, 20889–20894. [Google Scholar] [CrossRef] [Green Version]
- Noble, B.; Abada, P.; Nunez-Iglesias, J.; Cannon, P.M. Recruitment of the Adaptor Protein 2 Complex by the Human Immunodeficiency Virus Type 2 Envelope Protein Is Necessary for High Levels of Virus Release. J. Virol. 2006, 80, 2924–2932. [Google Scholar] [CrossRef]
- Hauser, H.; A Lopez, L.; Yang, S.J.; E Oldenburg, J.; Exline, C.M.; Guatelli, J.C.; Cannon, P.M. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 2010, 7, 51. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Shingai, M.; Welbourn, S.; Martin, M.A.; Borrego, P.; Taveira, N.; Strebel, K. Antagonism of BST-2/Tetherin Is a Conserved Function of the Env Glycoprotein of Primary HIV-2 Isolates. J. Virol. 2016, 90, 11062–11074. [Google Scholar] [CrossRef] [Green Version]
- González-Hernández, M.; Hoffmann, M.; Brinkmann, C.; Nehls, J.; Winkler, M.; Schindler, M.; Pöhlmann, S. A GXXXA Motif in the Transmembrane Domain of the Ebola Virus Glycoprotein Is Required for Tetherin Antagonism. J. Virol. 2018, 92, e00403-18. [Google Scholar] [CrossRef]
- Nishimura, Y.; Martin, M.A. Of Mice, Macaques, and Men: Broadly Neutralizing Antibody Immunotherapy for HIV-1. Cell Host Microbe 2017, 22, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Akkina, R. TRIM5alpharh expression restricts HIV-1 infection in lentiviral vector-transduced CD34+-cell-derived macrophages. Mol. Ther. 2005, 12, 687–696. [Google Scholar] [CrossRef]
- Chan, E.; Towers, G.J.; Qasim, W. Gene Therapy Strategies to Exploit TRIM Derived Restriction Factors against HIV-1. Viruses 2014, 6, 243–263. [Google Scholar] [CrossRef] [Green Version]
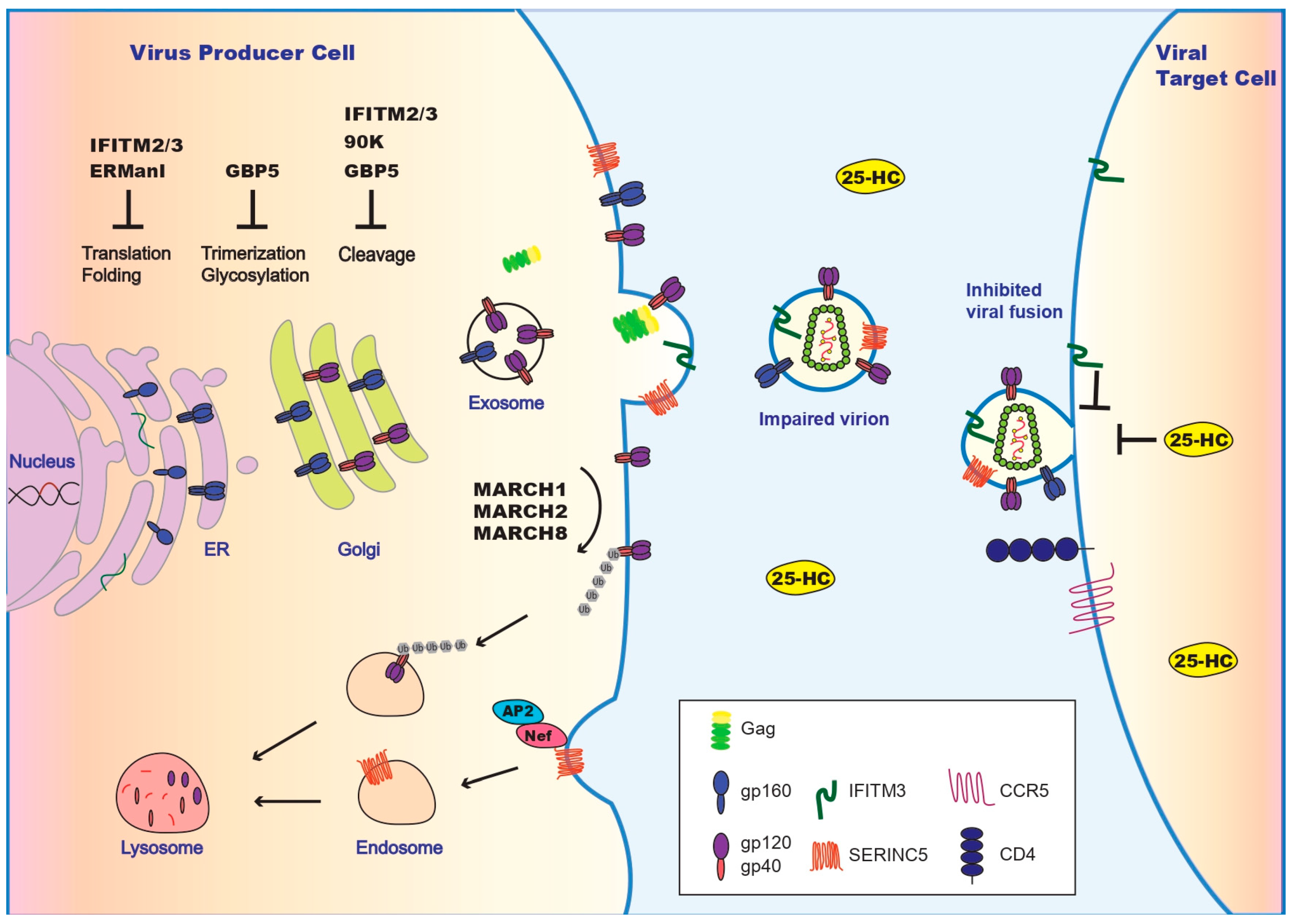
Figure 1.
Inhibition of HIV-1 entry by restriction factors and viral counter measures. Illustrated are restriction factors that operate in virus producer cells and inhibit Env synthesis at the endoplasmic reticulum (ER) (by IFITM2/IFITM3 and ERManl), impair Env maturation at Golgi (by IFITM2/IFITM3, GBP5, and 90K), and downregulate Env at the plasma membrane (by MARCH1/MRCH2/MARCH8). IFITM2/IFITM3 and SERINC5 get incorporated into HIV-1 particles and impair viral membrane fusion. In virus target cells, IFITM2/IFITM3 and 25-HC deter viral entry. HIV-1 uses Nef to downregulate SERINC5. The other viral countering strategies are summarized in Table 1. A more comprehensive illustration of antiviral restriction factors is presented in [19].
Figure 1.
Inhibition of HIV-1 entry by restriction factors and viral counter measures. Illustrated are restriction factors that operate in virus producer cells and inhibit Env synthesis at the endoplasmic reticulum (ER) (by IFITM2/IFITM3 and ERManl), impair Env maturation at Golgi (by IFITM2/IFITM3, GBP5, and 90K), and downregulate Env at the plasma membrane (by MARCH1/MRCH2/MARCH8). IFITM2/IFITM3 and SERINC5 get incorporated into HIV-1 particles and impair viral membrane fusion. In virus target cells, IFITM2/IFITM3 and 25-HC deter viral entry. HIV-1 uses Nef to downregulate SERINC5. The other viral countering strategies are summarized in Table 1. A more comprehensive illustration of antiviral restriction factors is presented in [19].


{kind=link}
Table 1.
Summary of the restriction factors that target HIV-1 Env.
| Restriction Factor | Impact on HIV-1 Env | Other Enveloped Viruses Affected | Virus Escape Mechanism | References |
|---|---|---|---|---|
| ErManI | Decrease Env expression via ERAD pathway; modulate glycosylation of HIV-1 Env | IAV | HIV Vpr increases Env expression | [24,26,27] |
| GBP5 | Impair cleavage of gp160; alter glycolysation of HIV-1 Env | MLV | Viral trade-off mechanism to increase Env expression by shutting down Vpu expression | [31,38] |
| 90K | Prevent gp160 processing; decrease mature gp120/gp41 in virions | EBOV | TBD * | [32,47] |
| IFITM2/3 | Deter viral entry into virus target cells; impair gp160 processing; promote gp120 shedding; decrease mature gp120/gp41 in virions; incorporate into virions and impair viral entry; | MLV, WNV, MPMV, EBOV, EBV, MeV, DENV | Overcome by HIV-1 Env | [33,61] |
| MARCH1/2/8 | Downregulate Env from the plasma membrane | HIV-2, SIV, MLV, VSV | TBD | [55,56,57] |
| SERINC5 | Impair virus infectivity; incorporate into virus particles; affect the conformation of the MPER region of Env | MLV, EIAV, EBOV | Downregulated by Nef from plasma membrane; countered by HIV-1 Env | [63,64,65,67] |
| 25-HC | Modify the secondary structure of the HIV-fusion peptide; prevents membrane fusion | VSV, ZIKV, EBOV, NiV, HCV, RVF | TBD | [83,84,85,91,92,93] |
* TBD: To Be Determined.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Beitari, S.; Wang, Y.; Liu, S.-L.; Liang, C. HIV-1 Envelope Glycoprotein at the Interface of Host Restriction and Virus Evasion. Viruses 2019, 11, 311. https://doi.org/10.3390/v11040311
AMA Style
Beitari S, Wang Y, Liu S-L, Liang C. HIV-1 Envelope Glycoprotein at the Interface of Host Restriction and Virus Evasion. Viruses. 2019; 11(4):311. https://doi.org/10.3390/v11040311
Chicago/Turabian StyleBeitari, Saina, Yimeng Wang, Shan-Lu Liu, and Chen Liang. 2019. "HIV-1 Envelope Glycoprotein at the Interface of Host Restriction and Virus Evasion" Viruses 11, no. 4: 311. https://doi.org/10.3390/v11040311
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.