Article Text

Abstract
Germline mutations in LKB1 cause the rare cancer prone disorder Peutz-Jeghers syndrome (PJS). Gastrointestinal hamartomatous polyps constitute the major phenotypic trait in PJS. Hamartomatous polyps arising in PJS patients are generally considered to lack premalignant potential although rare neoplastic changes in these polyps and an increased gastrointestinal cancer risk in PJS are well documented. These conflicting observations are resolved in the current hypothesis by providing a unifying explanation for these contrasting features of PJS polyposis. We postulate that a genetic predisposition to epithelial prolapse underlies the formation of the polyps associated with PJS. Conventional sporadic adenomas arising in PJS patients will similarly show mucosal prolapse and carry the associated histological features.
- LOH, loss of heterozygosity
- PJS, Peutz-Jeghers syndrome
- Peutz-Jeghers syndrome
- hamartoma
- prolapse
Statistics from Altmetric.com
SUMMARY
Germline mutations in LKB1 cause the rare cancer prone disorder Peutz-Jeghers syndrome (PJS). LKB1 can induce complete cellular polarity in single intestinal epithelial cells. Gastrointestinal hamartomatous polyps constitute the major phenotypic trait in PJS. Hamartomatous polyps arising in PJS patients are generally considered to lack premalignant potential although rare neoplastic changes in these polyps and an increased gastrointestinal cancer risk in PJS are well documented. These conflicting observations on the premalignant potential of PJS polyps frustrate the development of evidence based cancer surveillance strategies in PJS patients. The current hypothesis resolves this dilemma by providing a unifying explanation for these contrasting features of PJS polyposis. Several manifestations of PJS, including histopathology of the polyps, are consistent with mucosal prolapse. Analysis of model systems designed to investigate the function of either LKB1 or the LKB1 interacting protein Par1 and its mammalian homologues demonstrates that perturbation of either protein results in consistent phenotypes of epithelial prolapse. We therefore postulate that a genetic predisposition to epithelial prolapse underlies the formation of the polyps associated with PJS. Conventional sporadic adenomas arising in PJS patients will similarly show mucosal prolapse and carry the associated histological features. This notion resolves a persistent dichotomy on the origin of malignant change in PJS polyposis and shifts the focus away from the hamartomatous polyp as the chief precancerous feature in PJS.
INTRODUCTION
Historically, analysis of familial cancer prone conditions has contributed greatly to the understanding of tumorigenic processes operating in a sporadic setting. PJS is an autosomal dominant hamartomatous polyposis syndrome characterised phenotypically by mucocutaneous pigmentation and the occurrence of hamartomatous polyps throughout the gastrointestinal tract.1,2 These patients have a well established increased cancer risk and many die from malignancies at relatively young age (that is, fifth to sixth decade of life).3 In PJS the spectrum of tumours is diverse, and particularly uncommon types of neoplasms outside the gastrointestinal tract can occur. Currently advised screening strategies involve upper endoscopy, colonoscopy, and small bowel series in order to remove gastrointestinal polyps in patients aged 12 years and upwards.4 Although gastrointestinal cancers develop in PJS, it remains unclear whether these neoplasms are related to the polyps and whether the polyps carry an inherent (pre)malignant potential.
“Although gastrointestinal cancers develop in PJS, it remains unclear whether these neoplasms are related to the polyps and whether the polyps carry an inherent (pre)malignant potential”
The nature of polyp formation in PJS is unknown. Identification of germline mutations in the LKB1 gene,5 as the genetic cause in the majority of PJS families, should provide clues about the pathogenesis of PJS polyposis. The LKB1 gene was mapped after the observation of allelic loss of its wild-type counterpart in a minority of polyps from one patient and subsequent linkage of this locus to PJS.5 The observation of allelic loss suggested that inactivation of the wild-type LKB1 allele is responsible for the clinical manifestations of PJS, most notably the polyposis phenotype. The frequency of loss of the wild-type allele (loss of heterozygosity (LOH)) in human PJS polyps remains unclear as the relatively high rate reported in the literature6,7 may be due in part to induced losses because of technical shortcomings of genetic analyses. Mouse models have shown that haploinsufficiency is sufficient for polyp development8,9,10,11 and, therefore, a second hit is not a prerequisite for polyp formation (fig 1A). One model reported LOH in a minority of murine polyps11 but this may have been secondary to, for example, infection. Nevertheless, observations on LOH rates in human PJS polyps implied that loss of a given tumour suppressor function could explain polyp formation. Moreover, the rare observation of adenomatous and carcinomatous change within hamartomatous polyps12–15 was taken as proof of a tumorigenic sequence parallel to the adenoma-carcinoma sequence first documented in familial adenomatous polyposis. Hence the concept of a so-called “hamartoma-carcinoma sequence” was born,6,16,17 despite limited evidence and the observation that the microscopy of these hamartomas does not resemble (pre)neoplastic growth.

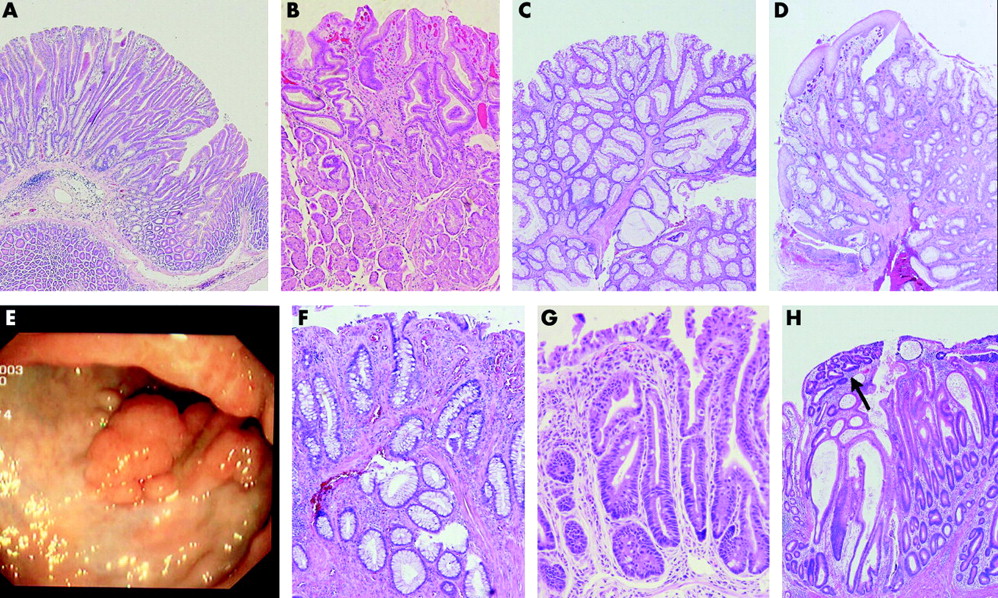
(A) Early gastric polyp in an Lkb1 +/− mouse, aged 29 weeks (Dr Mark Taketo). (B) Early gastric polyp in a patient with Peutz-Jeghers syndrome (PJS) (Johns Hopkins Medical Institution). (C) Early colonic polyp in a PJS patient (Academic Medical Centre). (D) Cloacogenic polyp (Academic Medical Centre). (E) Colonoscopy of a patient initially erroneously considered as PJS and reclassified as mucosal prolapse syndrome (LKB1 is wild-type in the germline). (F) Microscopy of the colonic polyp from (E). (G) Mucosal prolapse in the colon of a Mark2 −/− mouse (Dr Piwnica-Worms). (H) Adenoma with focally high grade dysplasia (arrow) and features of mucosal prolapse.
MICROSCOPIC CONSIDERATIONS
The histopathology of PJS polyps is distinct. Most are pedunculated with a mucosal surface that comprises the epithelial components typical for that particular site of the gastrointestinal tract. The crypts or pits as well as the deeper glands are often elongated, and the mucosa is typically draped on a core of arborising muscle bundles extending from the stalk into the head of the polyp (fig 1B, C). Although distinctive, this microscopy is not completely specific for PJS polyps. The pathologist can only confidently diagnose a PJS polyp if the patient has PJS or comes from a well established PJS family. Otherwise, strictly speaking, the histopathology report should read: polyp consistent with PJS polyp.
Mucosal prolapse is a lesion or phenomenon that is in the differential diagnosis of a PJS polyp. It can occur at different sites and settings in the gastrointestinal tract and raises various rare diagnoses, such as mucosal prolapse syndrome, cloacogenic polyp, and solitary rectal ulcer syndrome. Similar to PJS polyps, these lesions are characterised by a central area of proliferating muscle fibres surrounded by normal or reactive mucosa believed to be secondary to prolapse or protrusion of the mucosa into the lumen of the gastrointestinal tract (fig 1D, E, F).
“Mucosal prolapse is a lesion or phenomenon that is in the differential diagnosis of a PJS polyp”
Another feature observed as a result of mucosal prolapse is epithelial misplacement into the stalk and submucosa and, interestingly, this is also a common finding in PJS polyps.18 This “pseudoinvasion” is due to mechanical forces acting on a polyp during gut motility, which may lead to mucosal herniation into the stalk or submucosa. This finding can cause a spurious diagnosis of malignancy, which may account for a number of cancers reported in PJS polyps. Microscopic similarities between mucosal prolapse and PJS polyps are also pertinent as many PJS patients first come to medical attention with blood loss and symptoms of invagination such as colicky bowel pain or intussusception, features indicative of mucosal prolapse.
GENETIC CONSIDERATIONS
Recently, it was shown that LKB1 induces cell polarity.19,20 Loss of cell polarity is a well established feature of neoplastic growth. Moreover, evidence suggests that loss of polarity is not a consequence but rather a discrete step in the tumorigenic process.21–23 This latter consideration begs the question as to whether the role of LKB1 in regulating cell polarity can explain the pathogenesis of PJS polyposis and by extension the tumour prone condition.
LKB1, and its evolutionary orthologue par4, belong to a family of PAR genes which constitute a genetic conserved core module that cooperatively directs the execution of a cellular polarity programme.24 Recent data from various groups convincingly show that the LKB1 and PAR1 proteins cooperate closely and constitute a kinase cascade in vivo.20,25–27 Consequently, the results of PAR1 manipulation in model systems are pertinent, potentially reflecting the effects of disruption of the LKB1/PAR1 polarity pathway. In mammalian systems, four homologues of the Drosophila and C elegans PAR1 constitute the family of MARK kinases (that is, MARK1, MARK2, MARK3, and MARK4). Transgenic model systems mutant for PAR1/MARK or PAR4/LKB1 have generated interesting data regarding the above reasoning on epithelial prolapse in PJS. In C elegans, a phenotype of vulvar prolapse was observed on PAR1 depletion28; in the follicular epithelium of Drosophila oocytes mutant for either par1 or lkb1, cells are displaced from their adjacent environment29; and in murine models of Mark2 knockouts and heterozygotes, established separately in the laboratories of Dr Darmon and Dr Piwnica-Worms, mutant mice present with an unexplained phenotype of colorectal prolapse,30,31 a lesion similar to a common presenting symptom in PJS patients (fig 1G).2 These observations of epithelial prolapse have been confirmed in mammalian cell culture systems designed to investigate polarity maintenance where, similarly, cells mutant for MARK2 were squeezed out of an epithelial monolayer.32 In conclusion, consistent phenotypes of epithelial and mucosal prolapse have been observed on disruption of either PAR4/LKB1 or PAR1/MARK in various model systems, in keeping with close functional and genetic interactions between these gene products.
The PAR1/MARK kinases are part of a larger kinase family of AMPK-like kinases.33 This latter family consists of 13 closely related members, including MARK1 through MARK4 and the AMP activated protein kinase (AMPK). LKB1 activates all but one of the 13 AMPK-like kinases in in vitro kinase assays, underscoring the biochemical conservation of this interaction between LKB1 and the AMPK-like kinases.26 Two recent studies have expanded on this link, showing that LKB1 inhibits mTor by activating AMPK.34,35 Signalling through mTor acts as a cellular switch regulating cellular proliferation with changing nutrient availability. mTor signalling is inhibited under conditions of low nutrients, such as low intracellular ATP levels. Following AMPK activation, mTor inhibition restricts ATP consuming anabolic processes such as protein translation and cell growth in favour of energy conservation. The data in the two aforementioned studies show that mTor activity is regulated by LKB1 through TSC2 after direct activation of AMPK.34,35 As TSC2 is the tumour suppressor mutated in the tuberous sclerosis complex and mTor activity is further regulated by PTEN, the tumour suppressor mutated in Cowden’s disease, the authors converge on the conclusion that three hamartoma syndromes (that is, PJS, tuberous sclerosis complex, and Cowden’s disease) are all characterised by dysregulation of mTor activity.34,35
Importantly, all biochemical data in these latter studies were gathered in LKB1 deficient cells from Lkb1 KO embryos. This is relevant as biochemical data are extrapolated to PJS polyps which, as mentioned above, are haploinsufficient with respect to LKB1 for polyp formation. This implies that the effects of LKB1 heterozygosity on mTor signalling—in contrast with LKB1 nullizygosity—have yet to be firmly established. Moreover, from a clinical perspective, other than the word hamartoma there are few similarities between these syndromes, making it appear artificial to group these hamartoma syndromes together. Cowden syndrome is characterised by an increased risk for breast and thyroid cancer. The classical hamartoma in this syndrome is a skin lesion (that is, trichilemmoma). Although these patients may have gastrointestinal polyps, their morphology is different from PJS and there is no gastrointestinal cancer risk.36 In tuberous sclerosis complex the lesions typically affect the brain and skin, less frequently hamartomatous abnormalities can be found in the heart, lung, and kidneys, but gastrointestinal manifestations are extremely rare and there is no increased gastrointestinal cancer risk.37 Lastly, biallelic deletion of either one of both AMPK isoforms in murine models produces mice which display none of the gastrointestinal effects displayed by Lkb1 heterozygous or Mark2 heterozygous and deficient mice.38,39 Furthermore, although AMPK knockout mice show signs of impaired glucose tolerance in line with the presumed role of AMPK as a sensor of cellular energy status, none of these endocrine effects has been associated with PJS. This experimental outcome is in stark contrast with the consistent phenotypes independently described from manipulation of the LKB1/MARK pathway. To date, the only studies reliably documenting genetic interactions between LKB1 and a postulated target in vivo are those between LKB1 and PAR1/MARK.20,24,25
TESTING THE HYPOTHESIS
The above analysis of consistent phenotypes that arise from mutations in either PAR4/LKB1 or PAR1/MARK provides a strong impetus to elaborate on the concept of epithelial prolapse underlying PJS polyposis. One can envision how collections of cells squeezed out of their direct environment initiate mucosal prolapse in the gut. Once the prolapse has acquired the size and configuration of a polyp, patients run the risk of invagination. Interestingly, polyp formation in other organs such as the nasopharynx/respiratory tract, gall bladder, and bladder are also reported in PJS,17,40,41 indicating a general pathophysiological mechanism operative in this disorder.
“Interestingly, polyp formation in other organs such as the nasopharynx/respiratory tract, gall bladder, and bladder are also reported in PJS, indicating a general pathophysiological mechanism operative in this disorder”
As the present hypothesis departs from the canonical view of polyp formation whereby accumulating genetic accidents drive polypogenesis, genetic alterations obligate for polyp formation will not be demonstrable in hamartomatous PJS polyps. The observed “haploinsufficiency” for polyp formation in murine models8,9,10 agrees with this concept. In fact, only close scrutiny of polyp formation and visualisation of epithelial prolapse in more sophisticated model systems will allow this hypothesis to be tested directly. In view of their transparency, zebra fish could encompass the ideal model system for these studies, providing repeated longitudinal in vivo analysis of target organs. Contributory evidence would entail demonstration of polyclonality of PJS polyps. This coupled with the fact that PJS polyposis has been described on several occasions in newborns42,43 argues against accumulation of genetic insults mediating polyp formation. Finally, the current hypothesis predicts that the other phenotypic hallmark in PJS (that is, the mucocutaneous pigmentation) is due to a germline disruption of a polarity regulating pathway.
A UNIFYING CONCEPT OF PJS POLYPOSIS
As a consequence of the general predisposition to mucosal prolapse in PJS patients, other irregularities in the intestinal mucosal lining, most notably microadenomas, will similarly be prone to mucosal prolapse. Prolapse of incipient adenomas will then result in a histology of dysplasia in a PJS polyp. Microscopic features of mucosal prolapse are regularly observed in conventional adenomatous polyps or carcinomas arisen in an adenomatous polyp (fig 1H). The rare observation of dysplasia or even carcinoma in a supposedly pre-existent hamartomatous polyp in combination with the known increased gastrointestinal cancer risk in PJS has led to the spurious notion of a unique “hamartoma-carcinoma sequence” operative in PJS. This misconception in the order of events in the formation of dysplastic PJS polyps has confounded research into cancer pathogenesis in PJS. Although loss of the remaining wild-type allele with complete disruption of LKB1 polarity function in the polyps of PJS patients may promote tumour formation accounting for the increased cancer risk, gastrointestinal tumour initiation will still adhere to conventionally driven pathways.
“The rare observation of dysplasia or even carcinoma in a supposedly pre-existent hamartomatous polyp in combination with the known increased gastrointestinal cancer risk in PJS has led to the spurious notion of a unique “hamartoma-carcinoma sequence” operative in PJS”
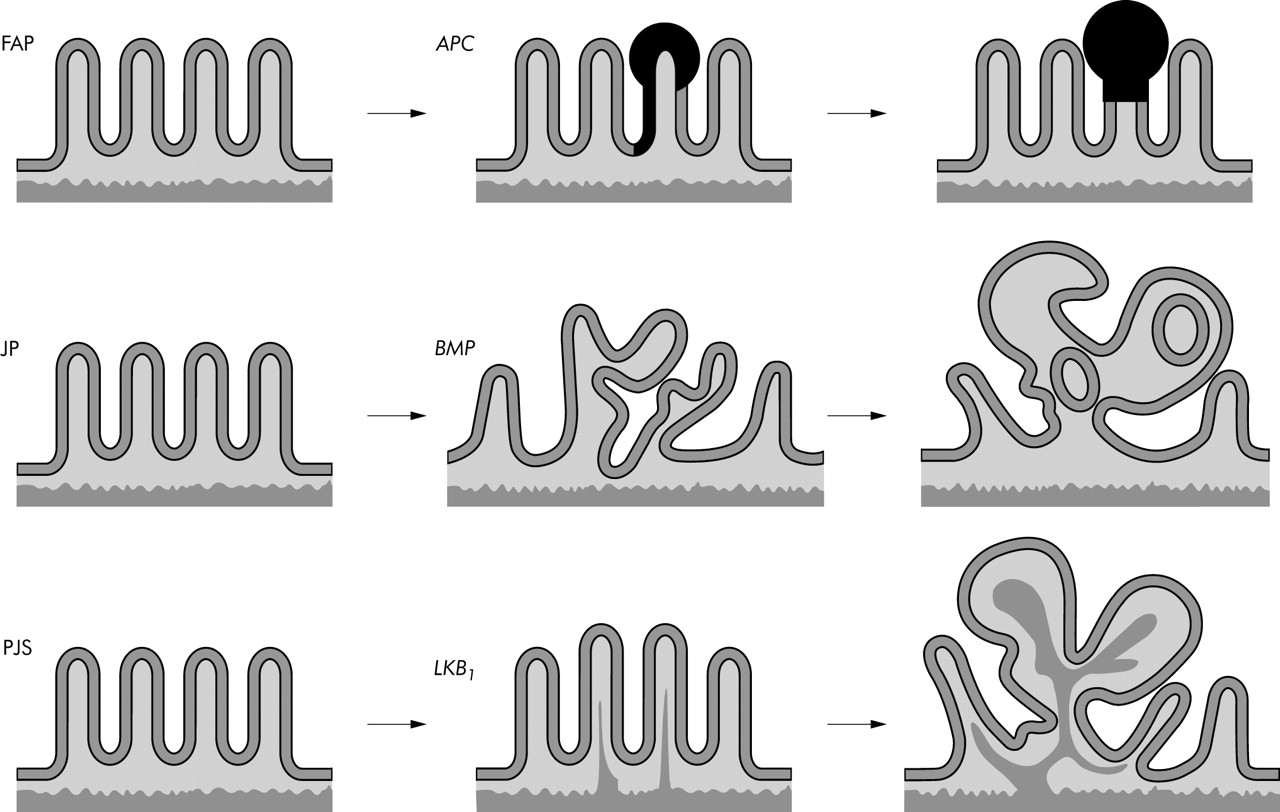
Interestingly, recent work on a mouse model of juvenile polyposis, the other gastrointestinal hamartomatous polyposis syndrome, indicated that in juvenile polyps, neoplastic growth also follows an adenoma-carcinoma sequence through Wnt activation (fig 2).44 This underscores the concept that colorectal cancers are initiated by disruption of a limited set of genetic pathways. The currently presented concept on the origin of malignant change in PJS polyposis reinforces this fundamental postulate.
{kind=link}
{kind=link}
Of the gastrointestinal polyposis syndromes, familial adenomatous polyposis (FAP), and the hamartomatous polyposis syndromes juvenile polyposis (JP) and Peutz-Jeghers syndrome (PJS) are the three main types. In all three syndromes the occurrence of multiple gastrointestinal polyps constitutes the phenotypic hallmark. In FAP disruption of the tumour suppressor gene APC leads to activation of the Wnt pathway, neoplastic proliferation, and clonal, expansion resulting in the formation of adenomas (upper panel). In JP, disruption of BMP signalling due to germline mutations in Smad4 or BMPR1A leads to a landscaper defect and crypts start to bud off or they grow perpendicular to the crypt-villus axis, resulting in the architectural changes typical for those observed in the hamartomatous polyps of JP (middle panel) (see Haramis and colleagues44). In PJS, a germline mutation of LKB1 is associated with loss of cell polarity function. This ultimately leads to epithelial misplacement and secondary changes due to mucosal prolapse (smooth muscle arborisation, pseudoinvasion) and formation of the “hamartomatous” polyps typical of PJS (lower panel). The polyps are non-neoplastic and therefore the hamartoma-carcinoma sequence does not exist (this manuscript).
IMPLICATIONS OF THE HYPOTHESIS
We postulate that hamartomatous polyp formation in PJS is the result of mucosal prolapse due to a germline disruption of a polarity pathway. This implies that the hamartomatous polyps observed in PJS are an epiphenomenon to the cancer prone condition and not obligate malignant precursors. The hamartoma-carcinoma sequence, as previously postulated, therefore, does not exist in PJS. If proven that the hamartomatous polyps carry no inherent risk of malignancy, our hypothesis bears immediate clinical relevance affecting surveillance strategies in PJS patients. PJS is the first human cancer prone condition in which the polyposis phenotype is linked to a germline disruption of a polarity pathway.
“The hamartomatous polyps observed in PJS are an epiphenomenon to the cancer prone condition and not obligate malignant precursors. The hamartoma-carcinoma sequence does not exist in PJS”
Note added in proof: It has recently been suggested that loss of polarity of function may lead to expansion of the stem cell pool by affecting asymmetric cell division.45 It is our belief that expansion of the stem cell compartment accompanied by mucosal prolapse may also be critical for the polyp formation in Peutz-Jeghers Syndrome and it can in addition contribute to cancer predisposition.
Acknowledgments
We thank Drs John Yardley, Guido Tytgat, Belur Bhagavan, Paul Drillenburg, and Anna-Pavlina Haramis for insightful discussions and critique, Drs Michel Darmon and Helen Piwnica-Worms for sharing information and material on unique mouse models, and Drs Josbert Keller and Ralph Carvalho for critically reading and discussing the manuscript.
Supported by a grant from the Netherlands Digestive Disease Foundation (MLDS #WS 01-03).
REFERENCES
Footnotes
-
Conflict of interest: None declared.
Linked Articles
- Correction