Article Text

Statistics from Altmetric.com
Non-erosive reflux disease (NERD) is defined as the presence of classic symptoms of gastro-oesophageal reflux disease (GORD) in the absence of oesophageal mucosal injury (or Barrett’s oesophagus) as determined by inspection at upper gastrointestinal endoscopy.1 As such it is regarded as being one of the two main phenotypes of GORD, the other being erosive oesophagitis (EO) where ulceration or erosions are evident. GORD is common, with estimates of 20–44% of Western populations having symptoms of GORD at least once a month and 20% weekly.2 The proportion of such patients with NERD is estimated to be between 503 and 70%.4 It is acknowledged that responses to standard acid suppressive treatments are 20–30% lower in patients with NERD than those with EO.5 Considering this, and the high prevalence of NERD, which is likely to increase in parallel with societal body mass index,6 the pathophysiological understanding of this condition remains a priority.
The concept of visceral hypersensitivity (VH) is now well established in a variety of overtly inflammatory as well as functional gastrointestinal conditions.7 ,8 This review introduces the molecular and physiological basis of VH, particularly in respect of acid-sensing receptors, and presents the evidence that VH plays an important role in the pathophysiology of NERD.
EO AND NERD: CONTINUUM OR DISCRETE CONDITIONS—THE ROLE OF VH
Within the spectrum of GORD, the pathophysiological relationship of EO and NERD remains the subject of debate.9 The classical and perhaps intuitive view that NERD is a mild form of GORD that might progress with time to EO is supported by some physiological, anatomical and histopathological findings. For instance, whilst demonstration of acid exposure is not a requirement for the definition of EO or NERD, there are studies that demonstrate that acid exposure as determined by 24 h pH studies is abnormal in only 45% of NERD patients compared with 75% of patients with EO, and that the mean recorded number of reflux events10 and extent of acid exposure11 ,12 are significantly lower in NERD. Similarly, the incidences of a variety of manometric physiological findings known to be associated with GORD, and the presence of hiatus hernia, are higher in EO patients than in those with NERD.9 ,13–15 Finally, whilst macroscopically normal, some attempts have been made to define “minimal change oesophagitis” in NERD using magnifying chromendoscopy with histological markers.16
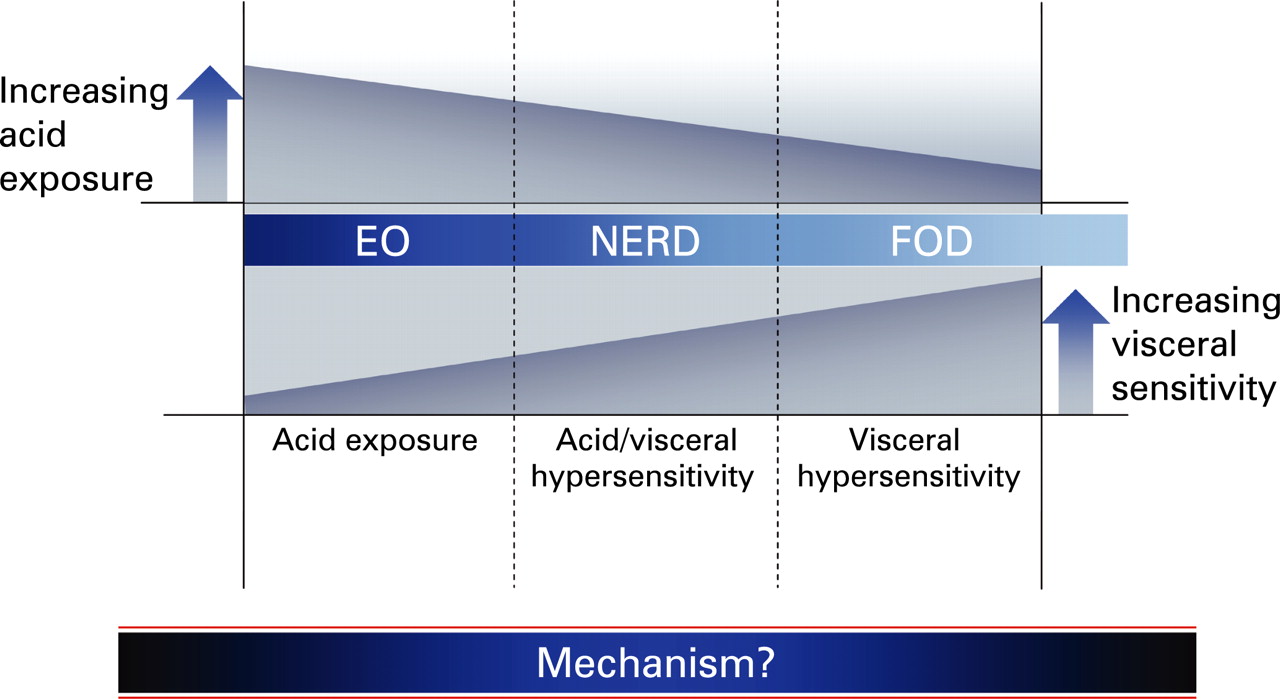
The assumption that EO and NERD represent one continuous disorder has, however, been challenged by studies that demonstrate some differing epidemiological features, responses to equivalent treatments (reviewed by Fass2) and perhaps the composition of refluxate in terms of acidity and gas,12 thus suggesting that NERD is at least in part a discrete disorder. Relevant to the discussion of whether this is true, or whether NERD fits into a continuum of acid-related disorders, are patients who have normal acid exposure and absent symptom–reflux association but who nevertheless have a clinical presentation almost inseparable from that of GORD. In current (Rome III) parlance, such functional oesophageal disorders (FODs: functional heartburn/chest pain of presumed oesophageal origin)17 may represent a further extension of this continuum. It is then plausible to suggest that the relationship between these variously defined disorders with homogeneous presenting symptoms follows a balance of increasing acid exposure as one progresses from FOD to EO (and perhaps further to Barrett’s). However, if differential acid exposure were the only factor, this could not explain how the clinical characteristics in terms of nature and severity of symptoms are almost equivalent in these groups.13 ,18 For this we need to suggest a further factor whose contribution to the pathophysiology of these disorders increases as one progresses in the opposite direction across the continuum (schematically represented in fig 1). Whilst more than one factor may account for this, the most compelling evidence to date exists for the role of the phenomenon known as visceral hypersensitivity (VH).
AN INTRODUCTION TO VH
Heightened perception of gastrointestinal sensation is termed VH and is commonly observed in patients with unexplained abdominal pain (where by definition it is termed visceral hyperalgesia). Numerous studies from the 1970s onwards, using a variety of intraluminal stimuli (usually mechanical or electrical), have demonstrated reduced pain thresholds in comparison with control subjects,19 ,20 and VH is now seen as the hallmark of functional gastrointestinal disorders (FGIDs).7
Extensive studies in man21 ,22 and experimental animals23 ,24 have attempted to unravel the respective contributions of organic and psychological mechanisms in the pathogenesis of VH in FGIDs. On the basis of some studies within the viscera and numerous studies in the peripheral somatic nervous system, it is clear that several mechanisms cooperate to varying degrees in the pathogenesis of VH. These include sensitisation of peripheral nerves (peripheral sensitisation), sensitisation of spinal cord dorsal horn neurons (central sensitisation) and “third party” interactions, predominantly via the so-called “psychoneuroimmune system”—that is, changes in cognitive and emotional functioning mediated by descending neural connections and the hypothalamo-pituitary axis (HPA), especially in response to stress.25 These mechanisms will be described in terms of basic science (in vitro and animal studies) before the evidence for VH is explored in human experimental studies of healthy volunteers and NERD.
VH: BASIC SCIENCE
Peripheral sensitisation
Nociceptors and primary hyperalgesia
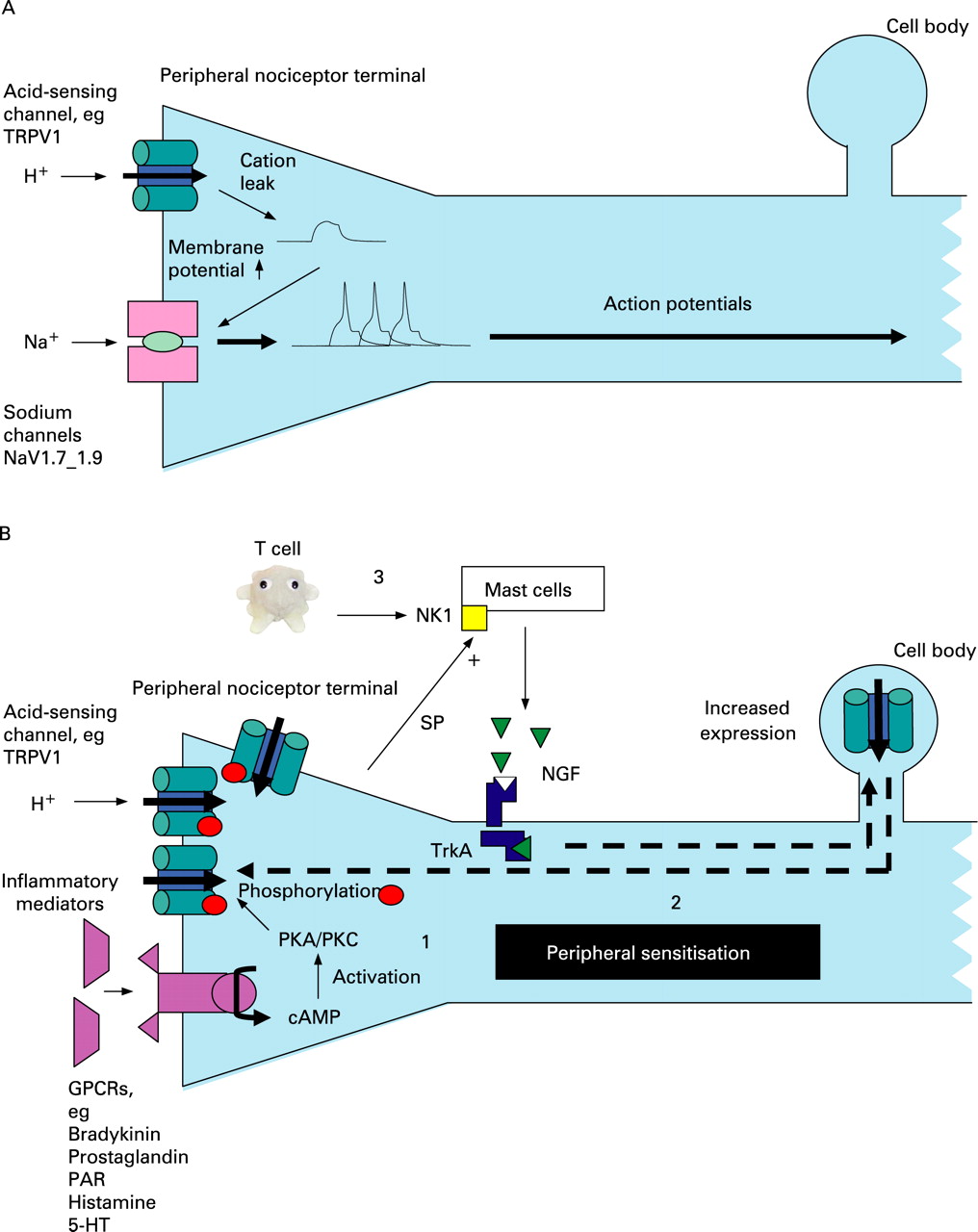
Extensive study of the peripheral somatic nervous system has elucidated many of the complex molecular events that underlie peripheral sensitisation. These are schematically represented in fig 2 and described here. In response to an excessive (noxious) stimulus or damage to tissue, several inflammatory mediators such as ATP, bradykinin, prostaglandins, histamine and H+ ions are released (in the gastrointestinal tract, the basolateral release of serotonin from enterochromaffin cells is also particularly important.26). These mediators have the common effect of reducing the transduction threshold of a variety of cation channels that are expressed on the peripheral terminals of specialised small myelinated (A delta-fibre) or unmyelinated (C-fibre) damage-sensing neurons that largely mediate pain (termed nociceptors).
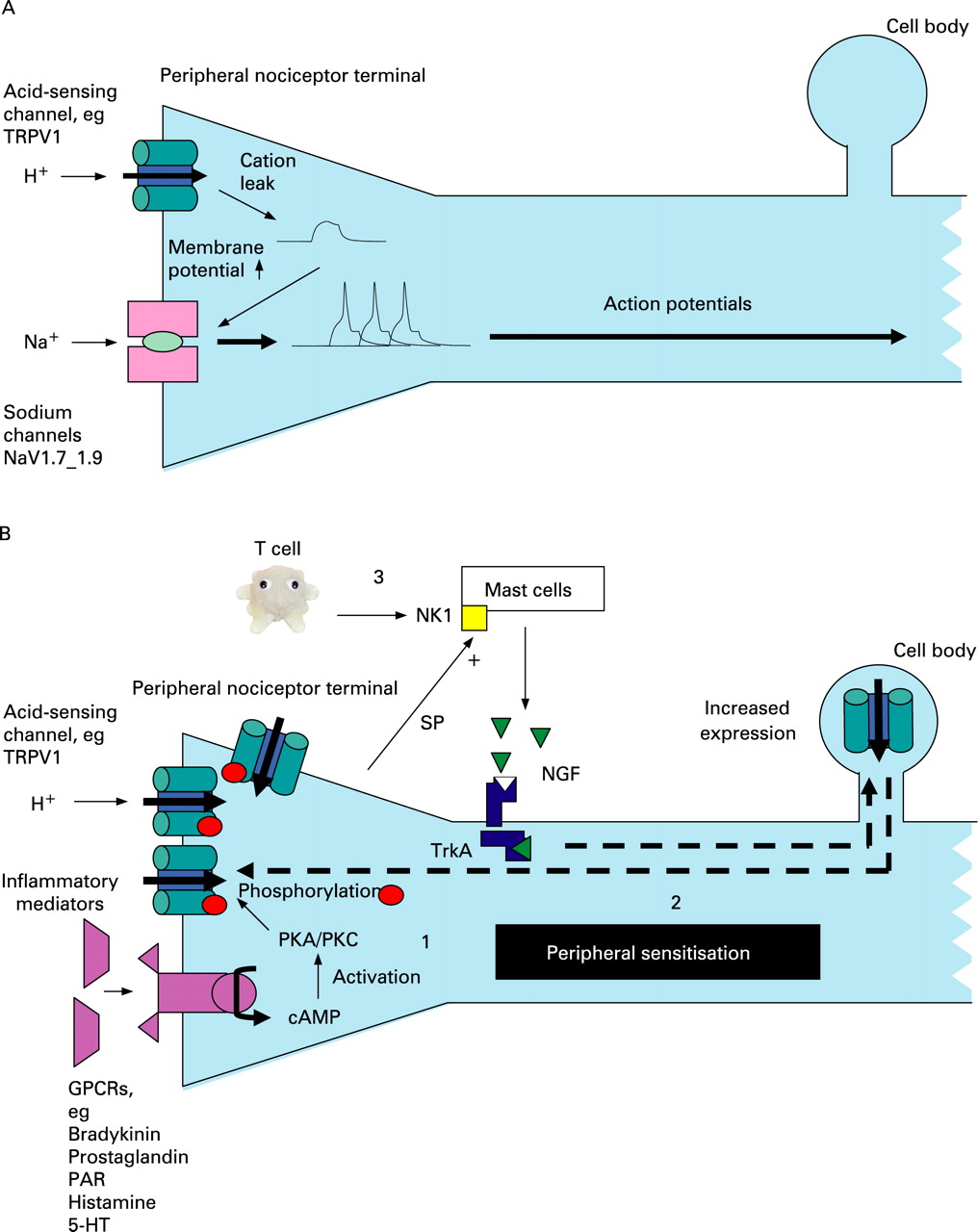
As in the skin, there are two broad classes of nociceptor based on differences in immunolabelling and trophic requirements.8 First are the so-called “peptide” nerves that contain neuropeptides such as substance-P and whose trophic requirement is nerve growth factor (NGF) acting on the tyrosine kinase receptor A. The second are those whose cell membrane contains the phospholipid label isolectin-B4 (IB4) and whose trophic requirement is glial cell line-derived neurotrophic factor (GDNF) acting via RET, a tyrosine kinase receptor encoded by the RET proto-oncogene (and so named after its method of discovery—REarranged during Transfection). Whilst some cation channels are found on both classes of nociceptor—for example, the transient receptor potential vanilloid receptor 1 (TRPV1)—others are preferentially expressed by a single class—for example, the purinergic receptor P2X3 on IB4-positive neurons. The final common pathway to an action potential and hence algesia in nociceptors is the activation of the sodium channels NaV1.7–1.9.27 ,28 These channels are sequentially activated by a reduction in the strongly negative (preserved by outward sodium pumping) resting membrane potential at the peripheral nerve terminal, and this can therefore occur when there is leakage of cations in response to inflammatory mediators including low pH (fig 2A). Sensitisation occurs when a persistently noxious stimulus leads to a variety of intracellular events that are shown in fig 2B (and legend) and discussed at least with reference to acid-sensing channels below. This peripheral sensitisation leads to the observed phenomenon of primary hyperalgesia (pain hypersensitivity at the site of injury).
Acid-sensing receptors in VH
Of relevance to acid-related disorders are several channels that are sensitive to low pH. These molecular acid sensors have been extensively reviewed elsewhere29 and include some classes of acid-sensing ion channels (ASICs), transient receptor potential (TRP) channels and ionotrophic purinergic (P2X) receptors. Whilst several other receptors may also have roles in acid sensing—for example, two pore domain potassium channels29 —these have been much less studied and are not reviewed further here.
ASICs (ASIC 1–3) are members of a voltage-insensitive, amiloride-sensitive epithelial Na+ channel/degenerin family of cation channels30 that are sensitive to pH ranges 6–7 and are considered to have roles in mechanotransduction. Whilst little is known of their role in inflammation/nociception in the gastrointestinal tract, their importance is suggested from altered transgenic animal responses to mucosal acid exposure31 ,32 as well as a study demonstrating their upregulation with gastrointestinal inflammation.33 NGF and serotonin (both of which are increased in mucosal inflammation) stimulate ASIC3 transcription in peripheral sensory neurons by a direct interaction with the promoter region of the ASIC3 gene.34
TRPV1 is one of a large family (n = 30 approximately) of highly conserved TRP channels that subserve sensory functions as diverse as hearing and pain.35 Of these, TRPV1 and TRPV4 respond to acidosis, even at modest pH changes (pH 6–7), and have important roles in pain mechanosensitivity. Acid binds to the extracellular domain of TRPV1, leading to inward currents that have been demonstrated in the ulcerated stomach.36 TRPV1 is regulated at a transcriptional level by a p38 mitogen-activated protein (MAP) kinase-dependent pathway37 that is activated by retrograde transport of NGF from the nerve terminal (bound to tyrosine kinase receptor A) to the dorsal root ganglion. NGF is produced by target tissues in response to inflammation and also by mast cells in response to substance-P binding to neurokinin 1 receptors.38 TRPV1 is expressed by both vagal and spinal afferents throughout the gastrointestinal tract of experimental animals39 and man.40 In a rat chronic reflux model, upregulation of TRPV1 protein has been demonstrated in the dorsal root and nodose ganglia41 in response to acid exposure, and TRPV1 antagonists ameliorate ulceration in this model.42 TRPV1 (and NGF) are also upregulated in human visceral inflammatory conditions40 ,43 and non-inflammatory VH.44
In common with many neuronal receptors, the transduction threshold of TRPV1 is reduced by phosphorylation induced by protein kinases A and C.45 These protein kinases are activated in response to injury by a cAMP-dependent mechanism and subserve the signalling from a variety of other G-protein-coupled receptors including those to phospholipids, 5-HT (5-hydroxytryptamine; a receptor that has been well implicated in VH throughout the gastrointestinal tract)26 and proteases (especially protease-activated receptor 2: PAR2).46 PAR2 may also be particularly relevant to VH in GORD since it is possible that in addition to acid, proteases can also sensitise, with pepsin demonstrated in the refluxate in some patients47 Proteases are also now believed to be important in the aetiology of VH in other FGIDs.48
P2X purinoceptors (P2X1–P2X7) are ligand-gated membrane cation channels that open when extracellular ATP is bound.49 Reduction in pH sensitises some P2X channels (especially P2X2) to ATP,50 and ATP has been found to sensitive vagal afferents to mechanical stimuli in the ferret oesophagus.51 P2X3 dorsal root ganglion/nodose protein expression is increased with chronic oesophageal acid exposure in a rat model,52 and increased peripheral P2X receptor levels have been demonstrated in the subepithelium of inflamed human bowel.53
Central sensitisation
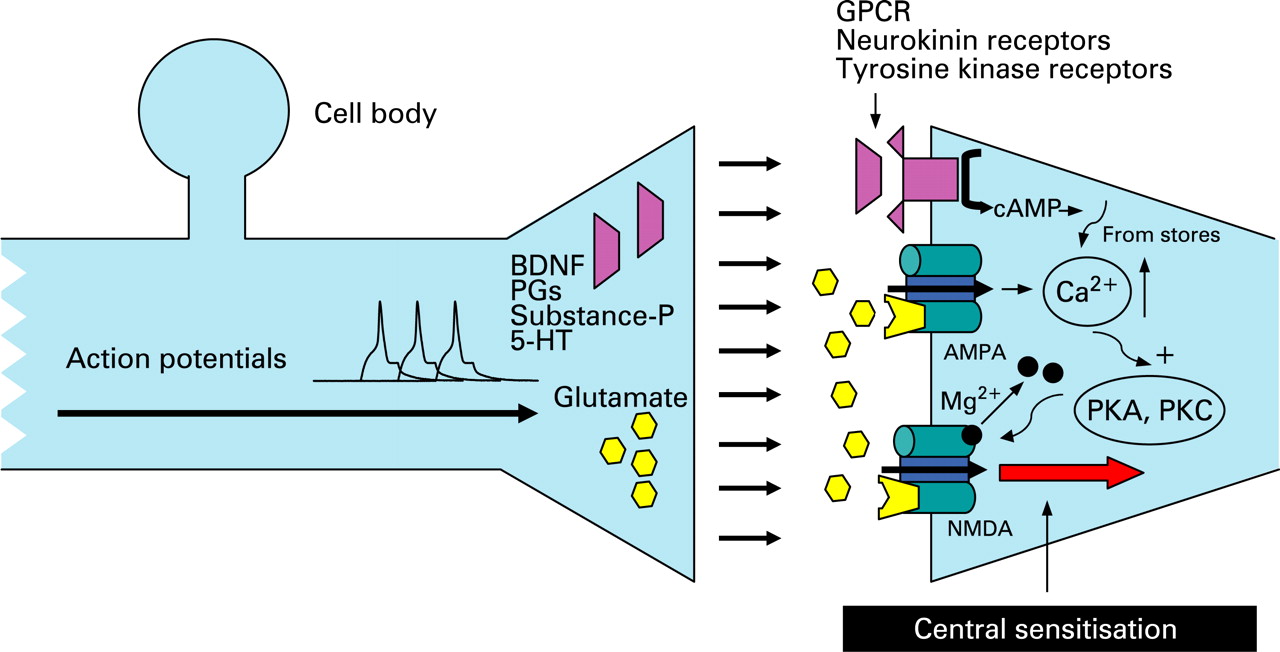
Repetitive firing of action potentials from the periphery also activates intracellular signalling cascades within the spinal dorsal horn neurons (fig 3). This leads to amplified responses to both noxious (hyperalgesia) and innocuous stimuli (allodynia).8 Such facilitation is triggered by the presynaptic release of several neurotransmitters including substance-P, glutamate and brain-derived neurotrophic factor (BDNF). Acting at their respective receptors, these lead to increased intracellular calcium and calcium-dependent activation of protein kinases A and C.54 This in turn leads to phosphorylation of N-methyl-d-aspartate (NMDA) receptors with a change in receptor kinetics that reduces their voltage-dependent magnesium block thus increasing subsequent responsiveness to glutamate.55 Central sensitisation also has effects on adjacent spinal neurons, leading to recruitment of previously “silent nociceptors” and hypersensitivity in areas (somatic and visceral) that are remote from the site of peripheral sensitisation (termed secondary hyperalgesia). Central sensitisation following noxious chemical stimulation with demonstration of viscerosomatic convergence has been shown in two studies of cats,56 ,57 and the role of NMDA receptors in this process was confirmed experimentally by their upregulation in a recent further cat model.58

Psychoneuroimmune interactions
The role of stress in the pathophysiology of gastrointestinal disorders has historically been regarded as separate from disease biology and classified under psychiatric co-morbidity. However, in the past decade, great strides have been made in understanding some of the basic concepts of brain–gut interactions and physiological and biochemical changes which occur during acute and chronic physical and psychological stress in humans. These are well reviewed elsewhere7 and are discussed only in the context of NERD below (with some relevant basic science).
Summary box
-
Visceral hypersensitivity (VH) is considered an important pathophysiological mechanism in functional gastrointestinal disease.
-
Three broad mechanisms are believed to underlie VH: peripheral sensitisation, central sensitisation and psychoneuroimmune interactions.
-
The molecules responsible for peripheral sensitisation include several acid-sensing cation channels, whose upregulation has been demonstrated in some animal models of GORD.
EXPERIMENTAL STUDIES IN HUMANS
Peripheral sensitisation
Demonstration of peripheral sensitisation in NERD
A number of studies have addressed the question of whether peripheral sensitisation, as defined by a reduction in the pain threshold to intraluminal stimulation at the site of acid exposure, is present in patients with GORD. Currently available stimuli include mechanical (usually balloon distension), electrical, thermal (hot and cold) and chemical. It should be noted that all have limitations in terms of either ensuring adequate mucosal contact (electrical and thermal) or allowing for confounding biomechanical changes in the viscus (mechanical), although these are less of a problem in the oesophagus compared with more distensible and geometrically complex viscera.59
In GORD, the presence of VH is well established to chemical stimulation and forms the basis of the Bernstein test in which 0.9% sodium chloride (control) and 0.1 M hydrochloric acid (HCl) (active) solutions are infused via a nasogastric tube in the distal oesophagus at a rate of 6–7.5 ml/min for up to 30 min or until oesophageal discomfort is induced. GORD patients demonstrate increased sensitivity to infusion60 ,61 although with poor correlation with symptom index as determined by 24 h pH monitoring.62 Mechanical sensitivity to balloon distension is found in general to be normal60 or paradoxically reduced in patients with proven EO.63 ,64 This is in contrast to patients with functional heartburn/chest pain, in which baseline mechanosensitivity65–67 and chemohypersensitivity68 ,69 have been demonstrated, and in which acid infusion leads to further increased sensitivity to subsequent electrical70 or mechanical stimulations.67 ,71 Hypersensitivity to heat (but not cold) has been demonstrated in patients with evident EO63 using a multimodal probe developed in Aalborg, Denmark. This probe facilitates multimodal sensory testing as well as incorporating impedance planimetry technology to address some of the concerns regarding biomechanical confounders.72 Robust normal ranges and preconditioning protocols73 exist for this device that has also been used in patients with NERD (see below).
Few studies have specifically selected patients with NERD defined on the basis of prior endoscopic findings. In comparison with patients with EO, recent studies demonstrate either almost identical responsiveness to acid infusion15 or increased acid sensitivity.74 ,75 These findings are consistent with a recent pH impedance manometric study that suggests that NERD patients are more sensitive to less acidic reflux.12 Only two studies to date have employed mechanical stimulation with variable results; the very recent findings of the Aalborg group using the multimodal probe demonstrating reduced mechanosensitivity and increased heat thermosensitivity (as in EO),76 being in contrast to increased mechanosensitivity as determined by patient-reported thresholds and cerebral evoked potentials in a Chinese study.77 A study of a mixed FOD/NERD group demonstrated increased electrical sensitivity at baseline and further increases after acid infusion.78 Thus results in NERD demonstrate equivalent or increased VH compared with EO, but perhaps less than in FOD. These findings are in keeping with the hypothesis presented earlier (and shown in fig 1)—that is, that hypersensitivity plays a greater role in FOD than NERD, and NERD > EO. These results are summarised in table 1.
Demonstration of peripheral sensitisation in human models of acid exposure
Several studies have induced VH in healthy volunteers after exposure of the distal oesopahgus to acid. In our model developed in Manchester and subsequently reproduced in Aalborg, a modest HCl (0.1–0.15 M) exposure of 30 min leads in most individuals studied to VH, as determined by a variety of stimuli including electrical,70 ,73 ,78–81 mechanical73 ,82 and thermal.73 In a recent study of cortical changes (measured by EEG) to experimental oesophageal acid sensitisation, the electrical stimulation electrode was placed endoscopically. This afforded the opportunity to visualise the mucosa at the site of infusion which had slight visible erythema but no erosions.81 Whilst clearly not a perfect model of NERD, these do at least prove that VH in response to acid exposure can occur in the presence of mucosal integrity.
ASICs in NERD
Upregulation of TRPV1 protein has been demonstrated in oesophageal mucosal biopsies of patients with EO83 and NERD,84 and mRNA levels of TRPV1 as determined by microarray have also been found to be elevated in mucosal biopsies from patients with NERD.85 The latter observation is of interest because it is generally accepted, not least from a basic cell biology stand point, that TRPV1 is transported to the nerve terminal as a protein,39 and an alternative explanation for observed mRNA increases may relate to increased numbers of inflammatory cells given that TRPV1 is now known to be expressed in leucocytes.86 To date, the roles of other acid-sensing receptors—for example, P2X and ASICs, have not been studied in man in health or after acid exposure. Furthermore, in contrast to other areas of the human gastrointestinal tract,33 ,37 ,41 ,42 the distribution of these receptors and TRPV1 has never been studied in full-thickness oesophageal tissue.
Mechanism of acid sensitisation in NERD
Clearly, the apparatus to sense (and become sensitised to) acid exists in visceral afferent neurons including the oesophagus. The question of how ASICs are actually exposed to reductions in pH is explicable in tissue acidosis such as occurs with inflammation but has been relatively unstudied in respect of luminal acidity. In all areas of the gastrointestinal tract, the terminals of both intrinsic and extrinsic neurons are located in the lamina propria (or deeper layers) and do not penetrate the epithelium, thus being protected from the hostile environment of the lumen, the widely held premise being that basolateral secretion of neuroactive substances—for example, serotonin 26—in response to luminal stimulation leads to activation of these endings. It is evident that ulceration/erosion of the epithelium as occurs in EO could expose these endings to H+ ions directly, but this is not the case in NERD. In the duodenum, the mucosal acid signal is transduced across the epithelium by diffusion of CO2, hydration to H+ and HCO3–, intracellular acidification, and exit of H+ via a basolateral sodium–proton exchanger (type 1).87 The oesophagus has a relatively thick (20–30 layers of) non-keratinised squamous epithelium, and even though this is penetrated to some extent by projections of the lamina propria, whether this mechanism operates in the oesophagus is speculative but unlikely. It is more probable that low grade inflammation plays a role in “transducing” the acid signal in NERD or that dilated intracellular spaces (DIS), known to be present in EO88 and NERD,89 ,90 permit some acid penetration. In respect of inflammation, studies of cytokines91–93 and of biopsies93 ,94 reveal that patients with NERD may have mild mucosal inflammatory changes not seen at endoscopy. The role of the immune system and possible important link to stress in mediating such changes are discussed sbelow.
Central sensitisation
Demonstration of central sensitisation in NERD
In patients with NERD, increased areas of visceral (upper oesophagus or stomach)79 ,82 and somatic hyperalgesia (chest wall)76 have been demonstrated, suggesting that like patients with functional oesophageal pain,70 central sensitisation plays an important role.
Demonstration of central sensitisation in human models of acid exposure
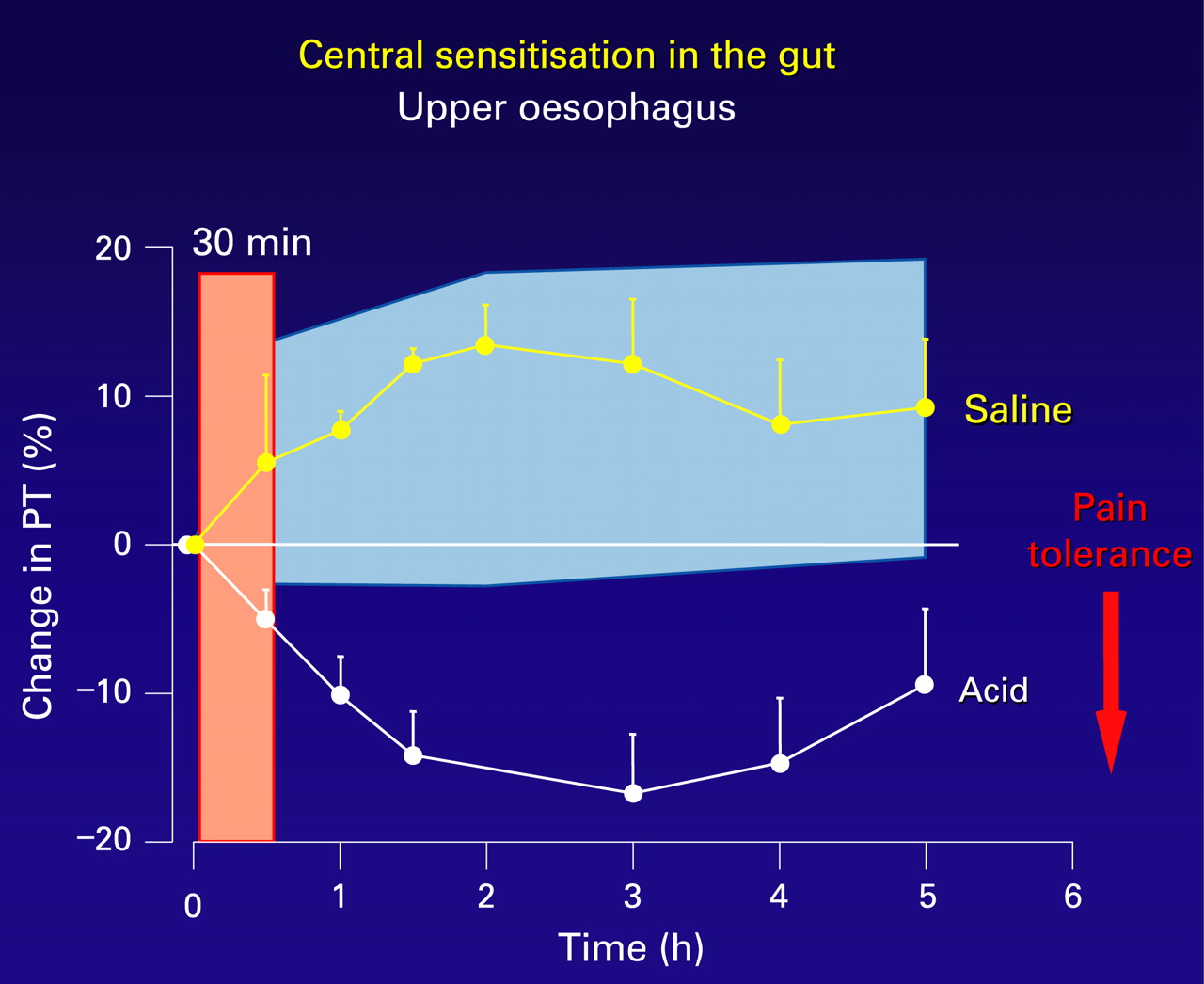
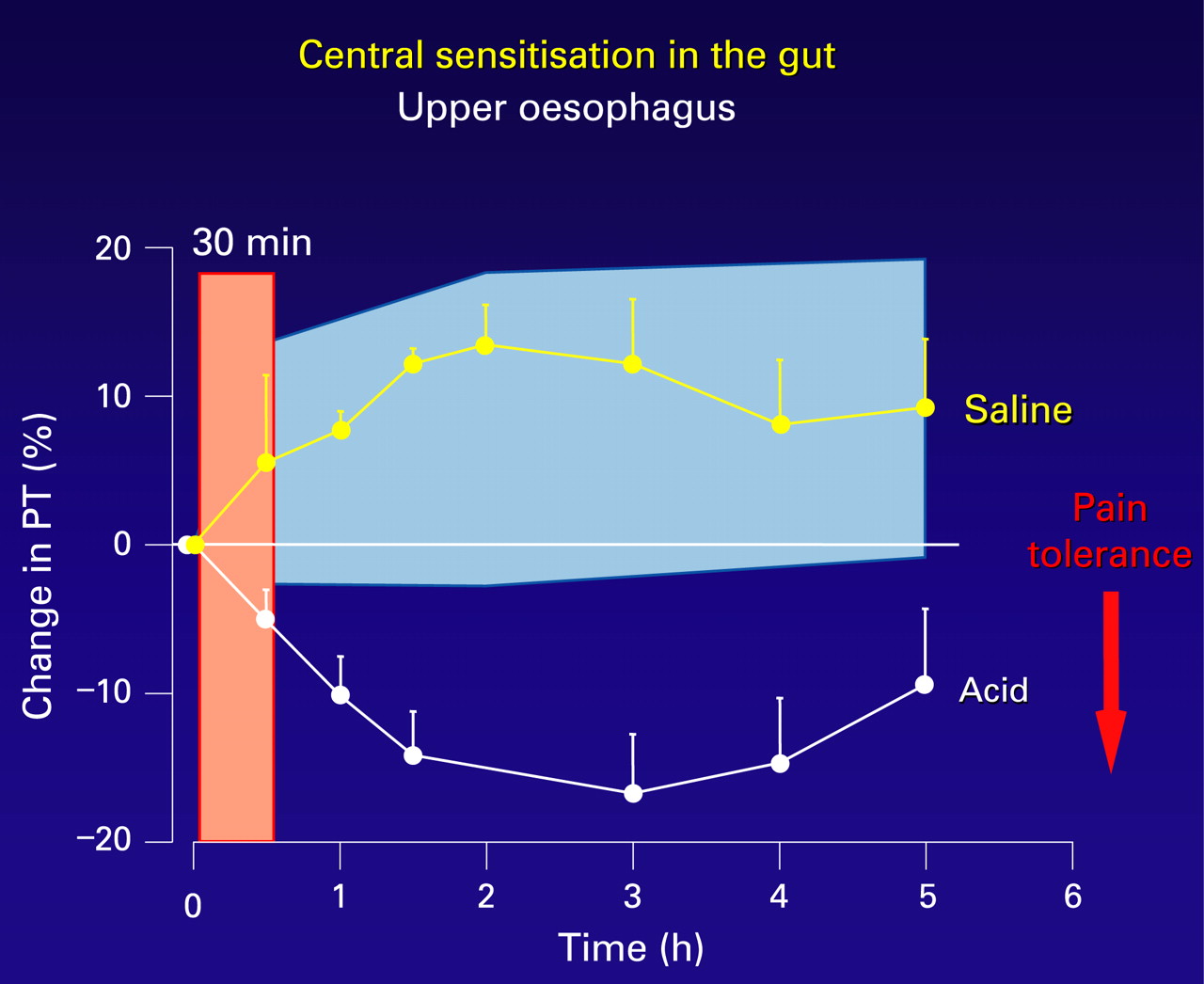
In the oesophagus, secondary hyperalgesia (viscerovisceral, proximal oesophagus; and viscero-somatic, chest wall) has been well demonstrated in a human volunteer model of distal oesophageal acidification (fig 4).70 ,73 This secondary hyperalgesia is attenuated by prostaglandin E2 receptor-1 (EP-1) receptor antagonism,78 and is both prevented and reversed with ketamine, an NMDA receptor antagonist,80 suggesting that central sensitisation not only is an important mechanism for the development of secondary hyperalgesia in this model but also that it occurs as in the somatic nervous system by a glutamate/NMDA pathway.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Psychoneuroimmune interactions
Psychological stress and NERD
In GORD, there is a general perception that psychological stresses and emotional perturbation induce or potentate GORD symptoms. In a Gallup poll, 64% of individuals with heartburn reported that stress increased their symptoms.95 Nevertheless, objective measurement of reflux using 24 h oesophageal pH monitoring demonstrated that <20% of patient-perceived reflux episodes were truly associated with acid in the oesophagus.96 This implies that even minor physiological noxious stimuli can be interpreted by the patient as a major symptom. Fass et al, using a dichotomous listening task, demonstrated that acute laboratory stress increased sensitivity to oesophageal acid exposure in patients with both erosive and non-erosive reflux disease.97 Relaxation training, in contrast, has been shown to decrease both heartburn symptoms and acid sensitivity in GORD patients.98 It is clear, therefore, that stress and psychological co-morbidity such as anxiety seem to play an important role in symptom generation in patients with GORD and, in particular, those with NERD.99 ,100 This emerging concept suggests that by modulating brain–gut interactions, symptom perception and possibly pathological events in the oesophagus of GORD patients might be altered.
As already discussed, in NERD, sensitisation of oesophageal sensory nerve endings might be facilitated either by low grade mucosal inflammation or by DIS, the latter leading to increased paracellular permeability and facilitation of acid exposure.88–90 It has recently been suggested that stress increases oesophageal permeability and dilates intercellular spaces to enhance exposure of the sensory nerve endings to the refluxate.101 Whilst acid with or without pepsin has proved sufficient to cause DIS in some studies in vivo102 and in vitro,89 a very recent rodent study from Leuven demonstrated that stress but not acid–pepsin exposure led to changes in permeability in vitro. This study demonstrated that when animals were stressed using partial restraint there was an increase in the submucosal mast cells as well as oesophageal permeability and DIS.103 This effect was potentiated when stress and acid–pepsin were administered together. The authors speculate that DIS-related increases in oesophageal permeability may be due to inflammation caused by mediators of mast cell degranulation, as has been shown in other parts of the gastrointestinal tract. Furthermore, mast cells express corticotrophin-releasing hormone (CRH) receptors104 and, recently, CRH receptor subtype 2 receptors have been identified in the rat oesophageal mucosa,105 suggesting that CRH and mast cells may be involved in regulating oesophageal secretomotor activity as well as modulating motility and other mucosal changes.25 These observations provide putative evidence for the role of the HPA axis in the pathophysiology of GORD.
Neuroimmune interactions in NERD
A full discussion of oesophageal mucosal immune interactions is beyond the remit of this review; however, the roles of such interactions are established in the pathogenesis of GORD, with activation of specific cytokines and chemokines well described.91–93 For instance, Fitzgerald et al reported a greater mucosal immune activation (interleukin 8 (IL8)) in patients with erosive oesophagitis in comparison with patients with non-inflamed or Barrett’s oesophagus.91 The role of inflammation in NERD was reviewed in 2000 by Hoshihara et al who have proposed a modification to the Los Angeles classification for oesophagitis.106 They have suggested that grade 0 (lack of erosions, suggestive of NERD) should be subdivided into grades M, which represents minimal change (irregular redness or whiteness) without any mucosal breaks, and grade N, which represents normal mucosa. Using this subdivision, Kanazawa et al compared immune activation in NERD patients with that in healthy controls,93 demonstrating that patients as a whole had higher IL8 levels than controls but that this was largely due to increases seen in patients with grade M, those with grade N and healthy controls having similar IL8 levels (suggesting that there is heterogeneity in NERD patients in terms of immune and inflammatory responses). They speculated that increased IL8 levels could lead to recruitment of neutrophils to the site of inflammation and cause oxidative stress, leading to tissue damage and thus also to exposure of nerve endings to nociceptive mediators. While the role of psychological stress in mediating oesophageal mucosal immune activation has not been explored in NERD, immune activation caused by mast cell degranulation, itself caused by stress, could be a possible mechanism,101 as has been demonstrated in other regions of the gastrointestinal tract.107
The autonomic nervous system in NERD
The extrinsic autonomic innervation of the oesophagus via parasympathetic vagal, and spinal afferent pathways plays an important role in modulating its sensory and motor function. It is therefore not surprising that investigators have examined the role of autonomic dysfunction in GORD,69 ,108 ,109 reporting a high incidence of autonomic abnormalities in patients with erosive oesophagitis with speculation as to a primary role for parasympathetic dysfunction. Recently, a lower resting vagal tone has been demonstrated in patients with erosive oesophagitis in comparison with NERD patients, suggesting that different degrees of vagal dysfunction may influence disease presentation in GORD.109 However, correlations between autonomic dysfunction and symptoms, acid exposure or psychological stress have not yet been demonstrated in GORD. Therefore, whilst a relationship between autonomic dysfunction and GORD seems to exist, the exact significance of this relationship still needs to be deciphered.
Summary box
-
Peripheral sensitisation has been demonstrated in patients with NERD and a healthy volunteer model of acid exposure. Upregulation of known acid-sensing receptors has been demonstrated in biopsies from patients with NERD. The mechanisms by which these receptors are exposed to acid still requires clarification.
-
Central sensitisation has been demonstrated in NERD patients and a healthy volunteer model of acid exposure, and is mediated by the same molecular pathways as in the somatic nervous system.
-
Psychoneuroimmune interactions are important in NERD and may explain alterations in acid permeability, mucosal immune activation and pain perception.
CONCLUSIONS
VH appears to be an important mechanism of symptom generation in patients with NERD. Evidence exists for peripheral and central sensitisation to acid (and possibly proteases) as well as brain–gut axis influences. NERD represents in many ways a greater management challenge than EO. It is probable, particularly in patients who fail to respond to standard acid-suppressing therapy, that new drugs will be found that modify not the exposure to acid but the process of sensitisation. Drugs modifying peripheral sensitisation—for example, TRPV1 antagonists—are already in phase III trials,110 and others—for example, PAR2 antagonists—are at a developmental stage. It is also likely that by studying in detail the stress-related effect on oesophageal function, additional therapeutic approaches may become evident. Further studies of the molecular changes that underlie VH in man and experimental animals will no doubt contribute to such exciting developments.
REFERENCES
Footnotes
-
Competing interests: None.
-
Funding: CHK is supported by HEFCE; QA is supported by a Medical Research Council Career Establishment Award and the Rosetrees Trust.