

Abstract
Objectives: To evaluate any association between the frequency of hereditary hemochromatosis (HFE) gene mutation (H63D and C282Y) and iron overload in beta-thalassemia major (BTM) patients.
Methods: The case-control study was conducted from June 2016 to February 2018. Blood samples from 204 BTM patients and 204 normal controls were taken from the Sundas Foundation Blood Bank.
These samples were analyzed for serum ferritin assay and HFE mutation. Ferritin level was measured on the ARCHITECT 1000SR. Both patient and control samples were analyzed for mutations using polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP).
Results: Serum ferritin levels for all patients were >1000ng/mL. The p.H63D mutation was observed in 23 (11.3%) cases, out of which 19 cases were heterozygous for p.H63D and 4 cases were homozygous. In control samples, 4 cases (2%) were found heterozygous for the p.H63D, and no homozygous mutation was found. Significantly high serum ferritin levels were found in BTM patients with the H63D mutation (p=0.00). In the case of p.C282Y, neither homozygous nor heterozygous mutation was found in patients or in controls.
Conclusion: H63D polymorphism is associated with iron overload in BTM patients. Larger-scale research is required to give an elaborated view of the association of the HFE mutation with iron overload in these patients and to confirm our conclusion.
Beta-thalassemia is a group of hereditary blood disorders characterized by anomalies in the synthesis of the beta chains of hemoglobin, resulting in variable phenotypes ranging from severe anemia to clinically asymptomatic conditions. The treatment strategy for beta thalassemia major (BTM) embraces blood transfusion, usage of antioxidants, induction of fetal hemoglobin, and hematopoietic stem-cell transplantation. However, the most common strategy of treatment is regular blood transfusion because it provides the body with a normal hemoglobin level, which is otherwise insufficient in these patients.1 Unfortunately, a number of complications are associated with regular blood transfusion, including iron overload and transfusion-transmitted viral infections like hepatitis B and hepatitis C.2 Hemochromatosis is a heterogenetic and autosomal recessive disorder of iron metabolism that is characterized by increased dietary iron uptake from the small intestine and its toxic accumulation in parenchymal cells of vital organs like the liver, pituitary, pancreas, heart and joints. Hereditary hemochromatosis (HH) is caused by a genetic mutation in the HFE, HJV, HAMP, TFR2, or SLC40A1 gene. In HH there is a constant absorption of iron within the upper part of the small intestine, hence increasing the total amount of iron in the body. This excess iron will ultimately start accumulating in various tissues because the body has no way to remove it. This depositing of iron in tissues leads to diseases including diabetes and cirrhosis. Although iron overload in transfusion-dependent thalassemia patients is generally due to repeated blood transfusion, there are additional factors that can increase iron absorption within the body, one of which is HH. Hereditary hemochromatosis may be co-inherited with beta-thalassemia and thus can increase the risk of iron overload, leading to numerous endocrine diseases, cardiac failure, hepatic cirrhosis, diabetes mellitus, and even death. Hereditary hemochromatosis due to mutations in the HFE gene is the most prevalent form of HH, and we studied this gene mutation. The most common HFE gene mutations are p.H63D and p.C282Y; p.H63D is relatively more prevalent than p.C282Y in South Asian countries including Pakistan, India, and Bangladesh. Data from South Asia shows that the prevalence of H63D mutation varies from 8%3 to 92%.4 In one Iranian study, an association between mutations in the HFE gene and iron overload in BTM was reported; specifically, patients carrying the H63D mutation had a significantly higher level of serum ferritin than those who did not show this mutation.5 The same association of H63D mutation has been shown in other studies as well.6-8 No association study has been conducted in Pakistan, but some studies in neighboring South Asian countries have also yielded the same results.4,9 Oliveira et al10 showed that the C282Y mutation worsens the clinical picture of beta-thalassemia patients in Brazil. Until now, no Pakistani group has studied the association between iron overload in BTM patients and C282Y and/or H63D mutation.
Methods
Our systematic search consisted of 4 keywords: 1) H63D mutation, 2) C282Y mutation, 3) HFE gene mutation AND hemochromatosis, and 4) beta-thalassemia major in Pakistan. We conducted a retrospective search of these keywords, with no specific time period, via PubMed and Google Scholar. All the articles were downloaded and viewed to short-list those articles that contained information about the association of C282Y and H63D mutations with iron overload in BTM patients.
This case-control study was conducted from June 2016 to February 2018 by following the principles of the Helsinki Declaration and was designed according to MOOSE reporting guidelines. Two hundred four patients and the same number of healthy blood donors (taken as control) were studied at the Sundas Blood Bank Thalassemia and Transfusion Center in Lahore, Pakistan. Beta-thalassemia major patients were already diagnosed, registered patients were being treated at the above transfusion center. Inclusion criteria encompassed patients with ferritin levels greater than 1000ng/mL, while all those patients were excluded who had hemoglobinopathy disorders other than BTM, had any other chronic illness, or were taking any hormonal therapy. All those blood donors were excluded who were suffering from any chronic disorder or taking any kind of hormonal therapy. In our study, more males were found than females among BTM patients as well as in the control group. The number of male patients at the Sundas Blood Bank was greater, and in the case of the control group, blood donors were mostly male. This parallels previously reported data in the literature, in which the male-to-female ratio was similar to that of our studied group.11,12 Our patient group consisted of mainly children, while our control group comprised mainly adults. This is because BTM patients usually die at a younger age and according to the WHO criteria of blood donation, donors should be adult. Additionally, to select children as a control was a difficult task due to sampling, the need for parental consent, and other related issues. Moreover, all these blood donors had normal serum ferritin levels, so they were chosen as a control group for our study.
This study was approved by the ethical board of the Sundas Blood Bank transfusion center.
Patient and control samples were analyzed for serum ferritin assay and HFE mutation. Samples were collected in above-mentioned transfusion center. The ferritin level of patients and donors (controls) were measured on the ARCHITECT 1000 SR. The ARCHITECT ferritin assay is a 2-step immunoassay used to determine the presence of ferritin in human serum and plasma using Chemiluminescent Microparticle Immunoassay (CMIA) technology. A prospective study was made for identifying subjects with hemochromatosis based on polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP). From this technique, controls and patients were screened for 2 known HFE gene mutations, Cys282Tyr and His63Asp. We used a primer set and the PCR-RFLP procedure according to Lynas, who describes this method as cost effective.13 After the PCR amplification, restriction enzymes were applied. The procedure for PCR-RFLP is given below.
First, DNA was purified from blood samples of both patients and controls using a spin protocol, following the QIAamp® DNA extraction kit method, and then analyzed qualitatively on 1% agarose gel. Polymerase chain reaction-RFLP was performed to analyze HFE gene mutations (C282Y and H63D). The primer pair for PCR amplification of C282Y mutation was C282Y forward (5′ TGGCAAGGGTAAACAGATCC 3′) and C282Y reverse (5′ CTCAGGCACTCCTCTCAACC 3′), and the primer pair for H63D mutation was H63D forward (5′ ACATGGTTAAGGCCTGTTGC 3′) and H63D reverse (5′ GCCACATCTGGCTTGAAATT 3′) as described by Lynas.13 Reaction conditions for both mutations were 1X PCR buffer with (NH4)2 SO4 (750 mM Tris HCl pH 8.8, 200 mM [NH4]2 SO4), 1 µM each forward and reverse primer, 5 mM dNTP mix, 2.5 mM MgCl2, 5 U/µl AmpliTaq DNA polymerase (Thermo Scientific) and 2 µl (~50 ng) DNA template. Polymerase chain reaction cycling conditions were as follows: initial denaturation at 95°C for 10 minutes, followed by 35 amplification cycles of denaturation at 95°C for 1 minute, annealing at 55.6°C (C282Y), 50.7°C (H63D) for 1 minute and extension at 72°C for 1 minute with a final extension at 72°C for 10 minutes. The PCR products were then digested with 5U of fast-digest enzyme RsaI and BclI (Thermo Scientific) for C282Y and H63D mutations respectively, followed by incubation at 37°C for 2 hours. After RFLP, electrophoretic separation was performed, and the digested fragments were run on 2% ethidium bromide–stained gel in 1X TBE buffer (85 mM Tris, 90 mM boric acid, 2 mM EDTA) at 95V for 2 hours, resulted bands were visualized and photographed.
Statistical analysis
After obtaining results, statistical data was analyzed by using the Statistical Package for Social Sciences Version 19 (Armonk, NY: IBM Corp.).14 Descriptive statistics were applied to calculate mean, standard deviation (SD), confidence interval, and minimum and maximum range, and analysis of variance (ANOVA) was applied to determine any significant differences between the means in each patient and control group mutation. P<0.05 was taken as the significance level.
Results
Patients’ mean age was 6.34 ± 2.27 (p=0.16). In contrast to the age of patients, the mean age of controls was 32.84 ± 3.91 (p=0.18). Gender statistics showed that 139 patients were male, representing 68% of the total sample, with a mean age of 6.23 ± 2.01. Females, of whom there were 65, constituted 32% of the sample size, with a mean age of 6.57 ± 2.75. In the control group, 161 of the donors were males and 43 were females, comprising 79% and 21%, respectively.
Table 1 shows that the mean level of ferritin in wild-type (non-mutated) patients was 2651.51 ± 1049.553ng/mL (mean ± SD), with a minimum value of 1081 and maximum of 9817. This mean value was lower than that of heterozygous HFE-mutated patients, which was 3566.53 ± 585.718 ng/mL, with a minimum value of 2350 and maximum of 4462. The mean ferritin level for homozygous HFE patients, which was 4517.50 ± 812.296ng/mL, with a minimum value of 3890 and maximum of 5710, was higher than in wild-type and heterozygous patients. There was a significant association found between the H63D mutation and iron overload in BTM (p=0.00).
Association of HFE mutations with serum ferritin in BTM patients and controls (n=204 each).
Serum ferritin levels of all control samples were in normal ranges, and the mean ferritin level of controls was far lower than that of the BTM patients. The mean ferritin level was 154.83 ± 30.487ng/mL with a minimum value of 90 and maximum of 250 in wild-type and 145.00 ± 35.355ng/mL with a minimum value of 120 and maximum of 170 in heterozygous mutation, and no control sample was identified with homozygous HFE mutation. No significant association was found between H63D mutation and iron overload in the control group (p=0.651).
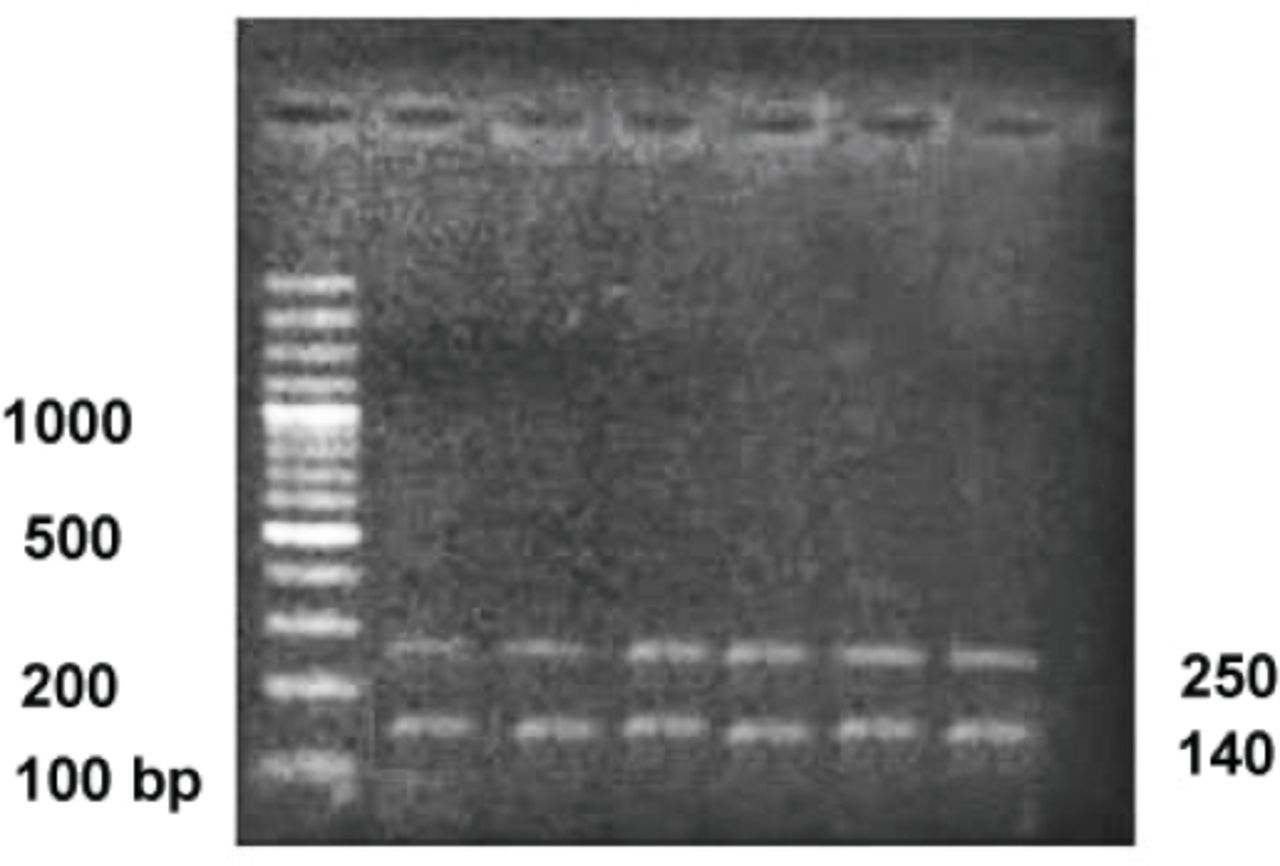
Polymerase chain reaction-RFLP of C282Y for all samples showed 2 wild-type DNA bands on agarose gel electrophoresis of size 250bp and 140bp13 (Figure 1). These results indicated the absence of this gene polymorphism in both patients and control samples. Polymerase chain reaction-RFLP analysis of H63D mutation resulted in 2 bands (138bp & 70bp), which represents wild type. HFE mutation was seen in 23 BTM patients. Nineteen of these 23 were heterozygous, with 208bp, 138bp, and 70bp bands and 4 samples were found homozygous for H63D mutation, having the 208 bp band only. These findings indicate 11.3% prevalence of H63D mutation in BTM patients in Pakistan. Four of the control samples exhibited p.H63D heterozygous mutation, but no homozygous mutation was found.

Restriction analysis of C282Y resolved on 2% ethidium bromide stained gel; Lane 1: 100 bp Ladder, lane 2-7: samples showing wild type DNA bands of 250bp and 140bp.
Figure 2 shows the bands of all 3 conditions on agarose gel.

{kind=link}
{kind=link}
Restriction analysis of H63D mutation resolved on 2% ethidium bromide stained gel; Lane 1: 100 bp ladder, lane 2 & 6; showing three bands of heterozygous mutation, lane 3; showing only one band of homozygous mutation, Lane 4, 5, 7 & 8; shows 2 bands depicting wild type DNA.
Discussion
The average age of patients involved in the experimental group was 6.34 ± 2.272 years, with a p-value of 0.18 (non-significant), which is comparable with data reported previously in Pakistan (10.1 ± 6.4 years15 and 11.18 ± 5.0716). This low average age is probably because of the typically low mortality age for BTM patients. The ratio of males to females in the current study was 2:1, indicating that males are more affected. The current study is consistent with previously reported data from different areas of Pakistan, including the data of Nawabshah17 who reported male-to-female ratios of 1.5:1, Karachi18 1.63:1, and Faisalabad19 1.9:1. Our results were also consistent with studies in the neighboring countries of India where the reported ratios of males to females were found to be 2.5:120 and Iran were 1.2:1.21
The mean age of the donors was 32.84 ± 3.915 years with a p-value of 0.18 (non-significant), which is also compatible with previously reported data for blood donors in Pakistan, wherein the mean age was shown to be 29.53 ± 7.034 years22 and 28 ± 13 years.23 The non-significant P-value of age for both control and patient groups means that there was no significant variation in individuals’ ages within each group, which is a good indication of precision in the results. Of the donors, 161 (79%) were male and 43 (21%) were female. This makes a male-to-female ratio of 3.7:1, which accords with previous data showing ratios of 3.8:111 and 2.52:1.12
The human body absorbs 1-2mg of iron from the diet, and an equal amount is removed via the gastrointestinal tract of the digestive system, yet the body is unable to secrete excess iron from the body. In beta-thalassemia patients, regular transfusions of 2 units of red blood cells are required (containing 200 - 250 mg of iron/unit), which can eventually result in the accumulation of more than 20g of iron in just 4 years of transfusion therapy, leading to hemochromatosis, a condition of excessive iron accumulation. Intracellular iron is stored in the form of the protein ferritin, with normal levels of <200 µg/L in females24 and <300µg/L in males. Our results showed that none of the patients had iron levels within the normal range. The mean ferritin level for all patients was 2773.32 ± 1071.908ng/mL, which is comparable with previous studies, where ferritin levels were established as 3391 ± 1960ng/mL,25 3103.9 ± 1747.4ng/mL,26 and 3348 ± 1794ng/mL.27 However, a few wild-type patients had higher ferritin levels than the patients with HFE mutation. Statistically, the p-value for association of ferritin with H63D mutation was significant (p=0.000), showing that the presence of this mutation has a consequence of elevated serum ferritin level. Serum ferritin levels in all healthy blood donors were within the normal range, with a mean value of 154.71 ± 30.453ng/mL. Only 2 individuals were found to be heterozygous for H63D mutation, with a p-value of 0.651 (non-significant), meaning that H63D mutation non-significantly correlates with iron overload in healthy persons, which is also consistent with previous data.28 Although the other common genetic cause of hemochromatosis is C282Y homozygosity, whose allele frequency is nearly 10% in the people of Northern Europe,29 this variation is present in very small numbers in the people of Asia. In the current study, no polymorphism of C282Y was identified in either normal or affected samples, which correlates with previous studies in which no C282Y mutation was reported in beta-thalassemia patients9,30 or normal persons.3,31
Among the BTM patients, the H63D mutation (homozygous and heterozygous) was found in 23 (11.27%). The partial transition of histidine with aspartic acids at codon 63 of the HFE gene was also found in 4 of the blood donors, which makes up only 1.96% (heterozygous only) of the sample size. These results are consistent with the findings of Enein et al,31 who conducted a mutational study within BTM patients. They found that 10% of patients of beta-thalassemia had the H63D mutation, which is comparable to our results, in which the allelic frequency of H63D was 11.3%, whereas 3% of their healthy controls also had the H63D mutation, which is also similar to our reported data. Our study is also consistent with an Indian study that showed 8.5% allele frequency of the H63D mutation in beta-thalassemia patients.9 However, some studies, such as one from Iran,32 argue that observed HFE mutation cannot be associated with iron overload in BTM. In short, there are 2 different points of view: some argue that H63D is significantly linked with the severity of iron overload in BTM patients, while others argue the opposite. But the current study showed a significant association of the H63D mutation with BTM (p=0.00).
Out of 23 cases of H63D mutation in patients, 83%19 were found to be heterozygous, and the remaining 17%,4 homozygous; similar results were found by Terzi et al33 who reported that 90% of the total H63D mutated patients had heterozygous H63D mutation, whereas 10% had homozygous H63D mutation. Similar results were also found in a study conducted by Ghadreri et al34 in this study, 12 patients of beta-thalassemia had H63D mutation, out of which 91.7% (n=11) were heterozygous and 8.33% (n=1) were homozygous.
Heterozygous H63D mutation was also found in 4 (1.96%) control samples, which is consistent with a study of Pakistani immigrants in the UK by Ali et al3 who found that only 8 out of 200 patients had the H63D mutation in heterozygous form.
In conclusion, the HFE gene mutation p.H63D is prevalent in Pakistan, and its prevalence supports the geographical spread of HFE gene mutations. However, C282Y is a rare mutation in Pakistan and neighboring countries, and our study found no presence of this mutation. The present study suggests that H63D is significantly associated with iron overload in BTM patients in Pakistan. Our results are consistent with most previous studies. But in these patients, iron overload may also be caused by multiple blood transfusions, hypersplenism, or other secondary non-HFE-linked hemochromatosis. Because of this uncertainty, hereditary hemochromatosis due to HFE gene mutations is difficult to study in Asian populations. Moreover, larger-scale research is required to give an elaborated view of the association of HFE mutation with iron overload in BTM patients.
Acknowledgment
We are highly indebted to Mr. Yasin Khan, Executive Director and Dr. Imran Qadeer, Laboratory Manager, Sundas Foundation (blood bank & thalassemia center), for their kind support and supervision in completing this work. We are thankful to all the patients and blood donors who participated in this research. We would also like to acknowledge “Scribendi” for providing us English editing service. This study is part of the Ph.D. thesis of Mr. Yasir Sharif.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received May 8, 2019.
- Accepted August 8, 2019.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.