


Beckwith-Wiedemann syndrome (BWS) is a disorder characterized by overgrowth, which manifests itself throughout prenatal and postnatal periods as macrosomia, macroglossia, and abdominal wall defects leading to omphalocele and diastasis recti. The clinical presentation of BWS (OMIM 130650) is markedly variable, and its multigenic and complex molecular causation is based on aberrant genomic imprinting. In most cases (85%), BWS is sporadic, but familial transmission accounts for 10-15% of the cases. Most of BWS pathogenesis is accounted for by epigenetic errors affecting the BWS-associated region on 11p15.5.1 CDKN1C is a negative regulator of cell proliferation, because it is a strong, tight-binding inhibitor of several G1 cyclin/CDK complexes. CDKN1C is 316 amino acids long, and it has 3 structurally distinct regions: a CDK-inhibitory domain, a domain that is rich in proline-alanine repeats, named the PAPA region, and a C-terminal conserved motif, named QT-box (Figure 1; Panel A).2 CDKN1C is preferentially transcribed from the maternal allele, and consequently, it is generally considered to be “paternally imprinted”.1

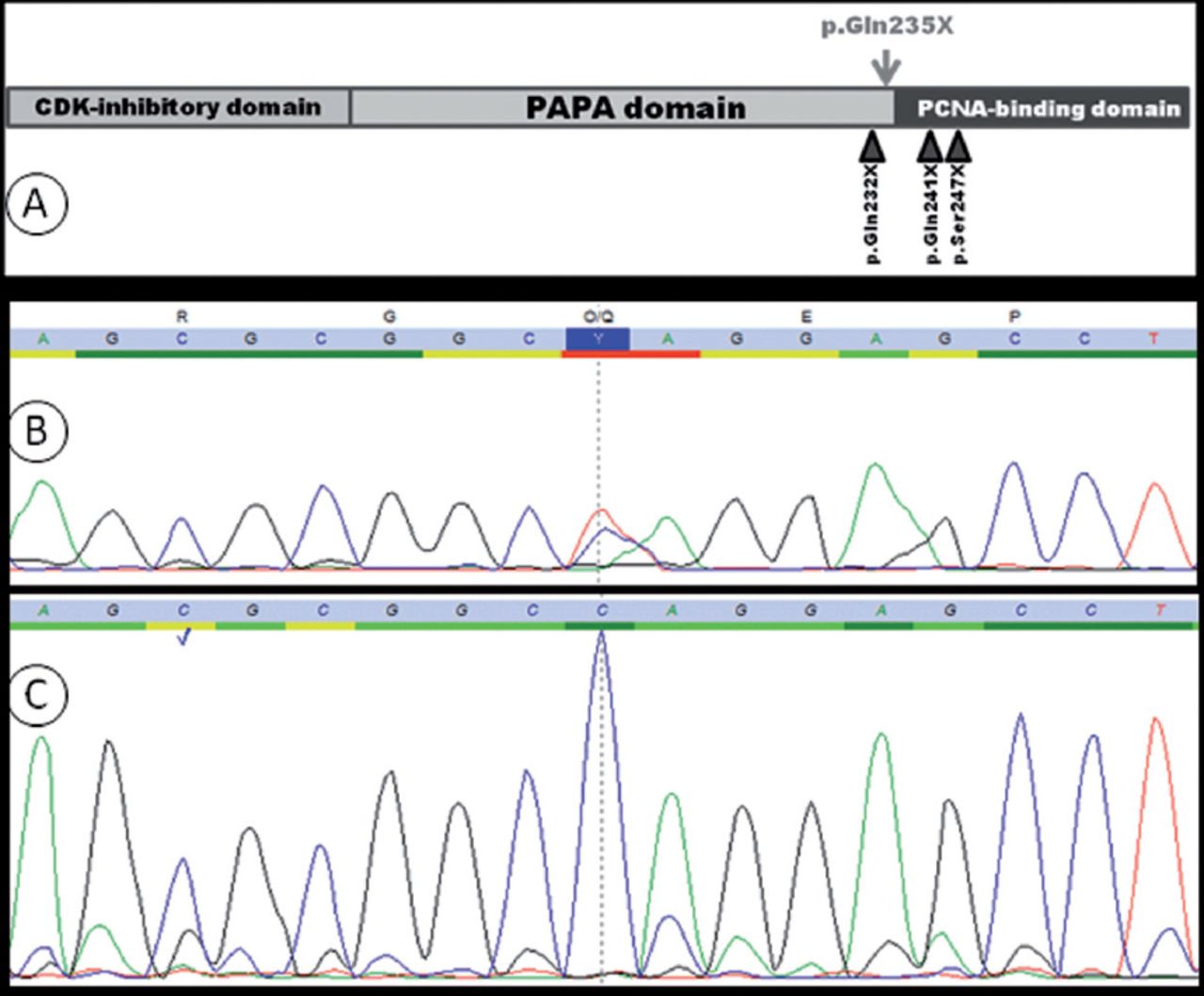
A graph showing: A) schematic representation of human CDKN1C. The position of the variant reported in this study is indicated with an arrow above the protein. B and C; sequence chromatograms of the mutated region in CDKN1C in the patient B) and his mother C). The patient harbors a heterozygous mutation (c.703C>T) in CDKN1C that results in (p.Gln235X), while the mother has only the WT version of this paternally imprinted gene.
In this study, we report a mutation in CDKN1C harbored by an Emirati BWS patient in a heterozygous state. The patient is a 9-year-old boy who was born at 37 weeks gestation by elective cesarean section for an omphalocele, which was detected antenatally. The parents are healthy and non-consanguineous. His Apgar score was 8 and 9 at one and 5 minutes, his weight was 4 kg, and his head circumference was 36 cm (both are above the 50th percentile).
Physical examination at birth revealed a dusky, distressed baby with dysmorphic features (including mid-face hypoplasia, infraorbital creases, facial nevus flammeus, macroglossia, posterior helical ear pit, large omphalocele (covered by Wharton jelly), and bilateral undescended testis. His systemic examination was unremarkable. The omphalocele was surgically repaired on the second day following birth; it contained small bowel, as well as small extra liver lobe. Echocardiogram of the patient revealed a small muscular ventricular septal defect, which closed spontaneously. His 6-monthly abdominal ultrasound revealed a non-progressive cyst in the left upper quadrant of the abdomen, as well as mild hepatomegaly. Levels of alpha-fetoprotein were repeatedly normal.
Upon informed consent, peripheral blood samples were collected from the affected child and his parents. Thereafter, genomic DNA was extracted from blood samples according to standard protocols. Exon2 and exon3 of CDKN1C were amplified by polymerase chain reaction and sequenced directly; these steps were carried out and validated at the Bioscientia Center for Human Genetics, Ingelheim, Germany. The consequence of the mutation unraveled in this study was assessed using ExPASy translation tool (http://web.expasy.org/translate/) and PolyPhen-2, which predicts the functional effects of human nonsynonymous SNPs (http://genetics.bwh.harvard.edu/pph2/). Sequencing showed a CDKN1C mutation; c.703C>T (p.Gln235X), in the patient, while no such mutation could be found in the parents (Figure 1; panels B and C). This variant affects codon 235, which is located just before the start of the PCNA-binding domain in CDKN1C, (Figure 1; panel A). The molecular alteration leads to creating a stop codon, and most probably, to the production of a truncated protein that completely lacks the QT-box. The C-terminal QT-box of CDKN1C can bind PCNA (proliferating cell nuclear antigen), which plays a crucial role in DNA replication.3 The high likelihood of producing a dysfunctional protein - as a result of the reported mutation - was corroborated by the prediction that PolyPhen-2 yielded for the variant as “probably damaging”.
The patient’s main BWS symptom was omphalocele, and interestingly; CDKN1C mutations appear to cause high frequency of omphalocele in patients.4 Additionally, our patient suffered cryptorchidism, which corroborates studies suggesting that genital anomalies were underreported previously.5 Although the patient showed no abnormal signs in relation to AFP tumor marker levels, it is imperative that he is followed up medically for any signs of neoplasia, as BWS patients are at a higher risk for developing tumors. In this context, it is particularly important to fully investigate the epigastric cyst that is uncovered by abdominal ultrasound. Such medical follow-up is recommended to continue until the age of 10 years. This study can help direct molecular investigations for the new cases that emerge locally and regionally, in order to elucidate the underlying causality properly and efficiently.
Footnotes
Disclosure. Authors have no conflict of interest, and the work was not supported or funded by any drug company.
- Received October 21, 2015.
- Accepted December 16, 2015.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.




{kind=link}