


Abstract
Hepatitis C virus (HCV) consists of envelope proteins, core proteins, and genome RNA. The structural genes and non-structural genes in the open reading frame of its genome encode functional proteins essential to viral life cycles, ranging from virus attachment to progeny virus secretion. After infection, the host cells suffer damage from virus-induced oxidative stress, steatosis, and activation of proto-oncogenes. Every process during the viral life cycle can be considered as targets for direct acting antivirals. However, protective immunity cannot be easily acquired for the volatility in HCV antigenic epitopes. Understanding its molecular characteristics, especially pathogenesis and targets the drugs act on, not only helps professionals to make optimal therapeutic decisions, but also helps clinicians who do not specialize in infectious diseases/hepatology to provide better management for patients. This review serves to provide an insight for clinicians and this might provide a possible solution for any possible collision.
The discovery of hepatitis C virus (HCV) can be traced back to the 1970s when Lacent first named a new kind of non-A and non-B viral hepatitis.1 Following the successful cloning of viral cDNA and sequencing of the full genome, the virus was officially named HCV and assigned into hepacivirus, flaviviridae.2 Approximately 170 million people worldwide are infected with HCV and it has been one of the major causes of viral hepatitis.3 When infected, this kind of viral hepatitis is more likely to end up in cirrhosis and hepatocellular carcinoma, both of which require proper management. Antiviral therapy is essential in treating chronic hepatitis C (CHC) and its stepping into a new era of direct-acting antivirals.4 Understanding its molecular characteristics, especially pathogenesis and targets the drugs act on, helps professionals to make optimal therapeutic decisions. It also helps clinicians who do not specialize in infectious diseases/hepatology to provide better management for patients as they might treat patients with HCV infection, even under antiviral treatment. Moreover, the development of HCV vaccine remains unsolved. Communications among clinicians from different specialties might provide intellectual collisions for possible solutions. Therefore, we intended to provide an insight into HCV characteristics for clinicians specialized in infectious diseases/hepatology or not, to serve all the mentioned purpose.
Article selection
Articles from Pubmed, Web of Science, Library, Information Science & Technology Abstracts (LISTA), and Library of Congress were selected with key words including HCV, molecular characteristics, life cycle, pathogenesis, and direct-acting antivirals. Articles in the last 5 years were preferred, along with classic articles in high-quality journals. The selected articles were categorized based on the abstract contents, and those deemed to be especially pertinent were read carefully. The major points of these articles were summarized in this review.
Molecular structure
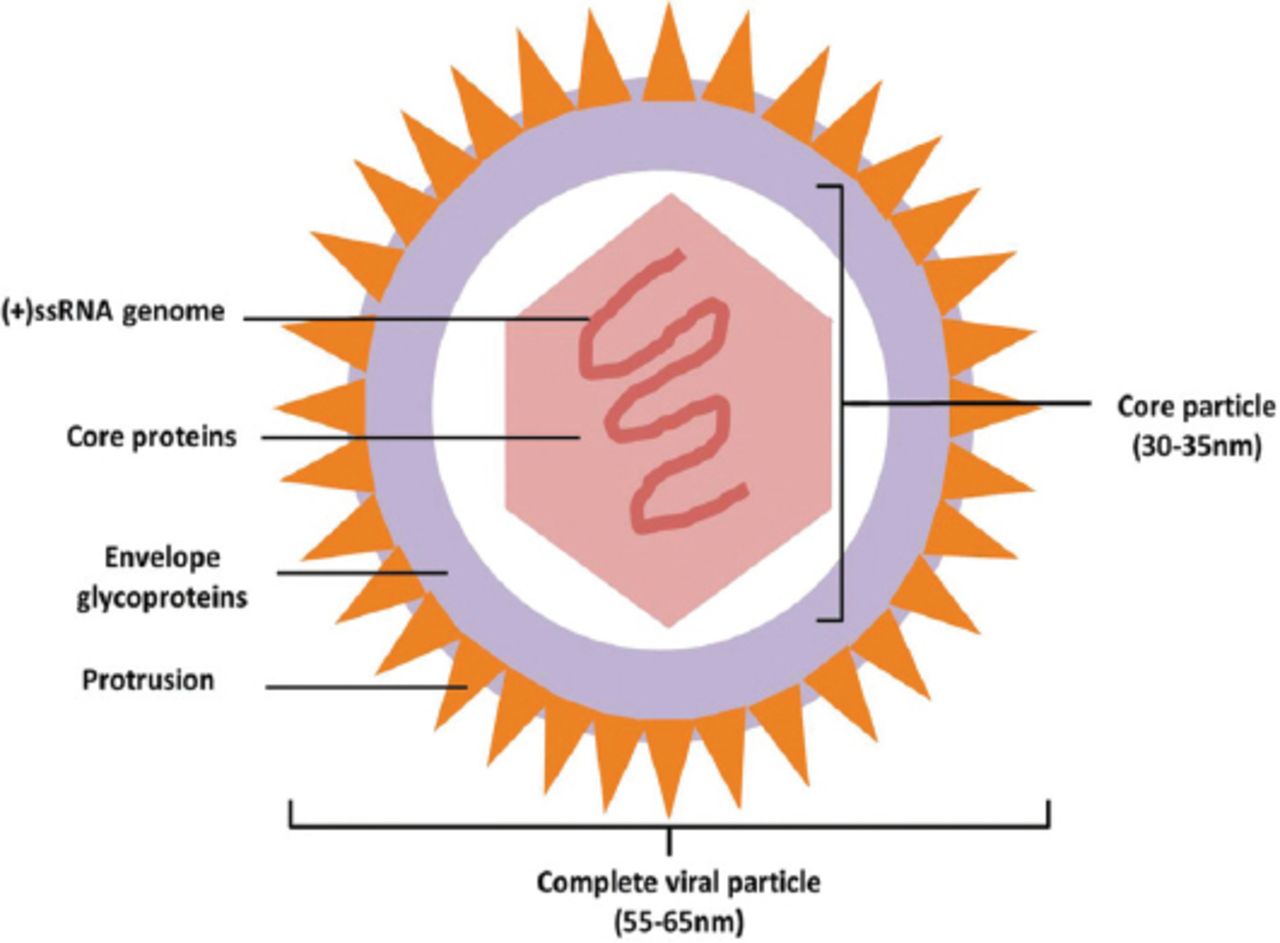
As the only liver-specific virus in hepacivirus, flaviviridae, the complete viral particle is approximately 55-65 nm in diameter. Outside the particle, there is a 7 nm-thick layer of envelope glycoproteins with 7 nm-long protrusions on it. The inside core particle is an icosahedron with a diameter of approximately 30-35 nm, consisting of core proteins and a single, plus stranded RNA genome in it (Figure 1). Aside from the complete particle, there are many nude core particles in HCV infected patients. After sucrose density gradient centrifugation, the harvested viral particles could be divided into 2 groups: the one with a high density of 1.186-1.213kg/L and the other with a low density of 1.099-1.127kg/L. The high-density particles are thought to result from the non-specific combination of viral particles and serum low-density lipoproteins or immunoglobulins. In vitro studies, the high-density particles showed impaired affinity and pathogenicity to susceptible cells, which might be related to the neutralization between attached proteins and cell receptors similar to antigens and antibodies.5

Ideograph of Hepatitis C virus (HCV) complete particle. The complete viral particle is approximately 55-65 nm in diameter. There is a 7 nm-thick layer of envelope glycoproteins outside the particle with 7 nm-long protrusions on it. The inside core particle is an icosahedron with a diameter of about 30-35 nm, consisting of core proteins and a single, plus stranded RNA genome in it. (+)ssRNA - single plus strand RNA
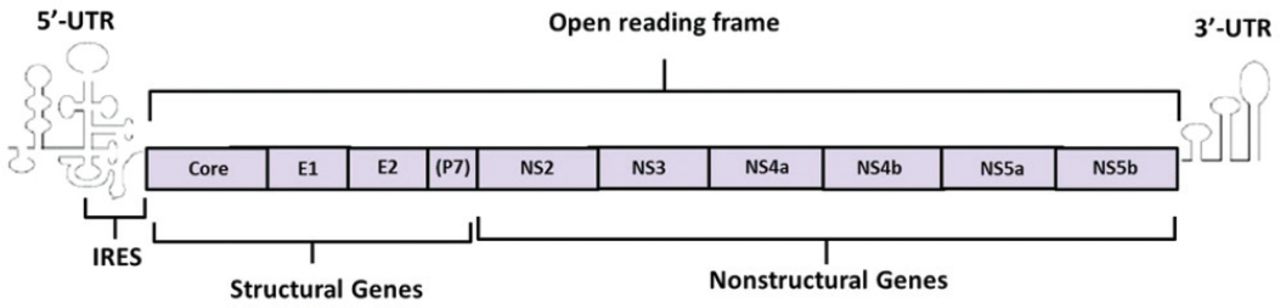
The HCV genome is a 9.6 kb in length. It consists of 2 untranslated region (UTR) at the 5’ and 3’ ends and an open reading frame (ORF) in the middle. There is an internal ribosome entry site (IRES) in the 5’-UTR, which can induce the initiation of viral protein translation. Internal ribosome entry site sequence is relatively conservative and could be the target for therapeutic ribozyme.6,7 Unlikely, the 3’-UTR mainly works on the regulation of virus replication.8 The ORF encodes a precursor polyprotein composed by 3010 amino acid residues. Afterwards, the precursor protein was cleaved by viral or host protease into more than 10 structural or non-structural proteins, which participated in the processes of virus replication, assembly, and infection. According to the encoded proteins’ functions, the ORF was divided into structural genes and non-structural genes. The core protein encoded by core region of the structural genes is a highly conservative protein containing several B cell epitopes with immunogenicity While the envelope region of structural genes encodes the glycosylated proteins of the envelope membrane including Envelope 1 (E1) and Envelope 2 (E2). These 2 proteins show high heterogeneity in different virus strains. The P7 protein, which is located on the end of E2 protein, is also encoded by the envelope region. It is a polypeptide resulting from incomplete enzymolysis of E2. On the other hand, there are 6 regions in the non-structural genes including NS2, NS3, NS4a, NS4b, NS5a, and NS5b. The NS2 protein forms the protease with the N-terminus of NS3 protein to modify the precursor polyprotein. In the C-terminus of the NS3 protein, there is a nucleoside triphosphate hydrolase (NTPase) domain and an RNA helicase domain involved in virus replication. These 2 domains could also be studied as possible targets for antiviral drugs. NS4 gene encodes 2 kinds of highly immunogenic proteins, NS4A and NS4B, which could be used for medical antibody development. In the genotype of the 1b virus strain, the NS5A encoded by NS5 gene contains a domain called interferon-sensitive determining region (ISDR), affecting the patients’ response to interferon therapy. The other protein NS5B encoded by NS5 gene plays the role of RNA-dependent RNA polymerase, which could be considered as a potential target for antiviral drugs as well9,10 (Figure 2).

Ideograph of Hepatitis C virus (HCV) RNA genome. The HCV genome consists of 2 UTR at the 5’ and 3’ ends and an open reading frame (ORF) in the middle. There is an IRES in the 5’-UTR inducing the initiation of viral protein translation. The 3’-UTR mainly works on the regulation of virus replication. The ORF encodes a precursor polyprotein composed by 3010 amino acid residues which could be cleaved by viral or host protease into more than ten structural or non-structural proteins. UTR - untranslated region, IRSE - internal ribosome entry site, (p7)- the encoding gene of P7 is a part of E2 gene.
The life cycle of HCV
In both in vitro and in vivo investigations, the HCV particles present a tendency to infect hepatocytes. By specifically attaching to entry factors, the viruses release their nucleic acids into the hepatocellular cytoplasm through the membrane. Then, the virus replicates according to the template viral RNA, which is then translated into glycoproteins. After modification, the glycoproteins are converted into mature viral proteins for assembly of progeny viral particles. Finally, the progeny viruses are released out through the secretion pathway of the host cells and start a new cycle of their life11 (Figure 3).

Ideograph of Hepatitis C virus (HCV) life-cycle. The life cycle starts from viral attachment to entry factors. Then, viruses release their nucleic acids into the hepatocellular cytoplasm through the membrane. Afterwards, the virus replicates according to the genome RNA, which is then translated into glycoproteins. With proper modification, the glycoproteins are converted into mature viral proteins for assembly of progeny viral particles. In the end, the progeny viruses are released out through the secretion pathway of the host cells and start a new cycle of their life. Protein translation: during translation, the hydrolysis of precursor poly-proteins occurs as well. Replication complex: the complex consists of ER, viral genome and nonstructural proteins including NS2, NS3/4A, NS4B, NS5A and NS5B.
Attachment and entry
At first, the mucopolysaccharide and low density lipoprotein (LDL) receptors on the host cell membrane mediate moderate attachment of viruses to cells.12,13 Then, the viral envelope glycoproteins combines with the entry factors specifically, initiating the host cell endocytosis to allow the viruses into the cytoplasm. As a member of the tetraspanin superfamily, CD81 is the definite receptor that mediates the viruses’ entry. The extracellular part of CD81 is formed by 89 amino acid residues. Such cyclic structure is the specific binding domain of envelope glycoprotein E2.14,15 In recent years, another entry factor scavenger receptor-BI (SR-BI) has been discovered. It also plays an important role in the virus entry because when anti-SR-BI or its siRNAs were introduced, virus infection to host cells could be inhibited effectively. The regulation on SR-BI expression could affect the susceptibility of host cells as well as the regulation of CD81 expression. However, because both lipoproteins and E2 could interact with SR-BI, it deserves further investigations in whether specific combination occurs with viral enveloped proteins or combined lipoproteins on the virus particles.16
Interestingly, some experts observed a phenomenon that even if the sustained expression of CD81 and SR-BI were detected in some cell lines, HCV could not effectively infected them. A series of cooperative factors were found when scholars delved into the mechanism. These proteins such as tight junction proteins claudin (CLDN)1,6,9 in CLDN family could form a receptor complex along with CD81.17,18 When the formation of the complex was inhibited by down regulating the activity of protein kinase A, the host cells were less susceptible to virus infection. Another tight junction protein (occludin) was also considered to be involved in the entry procedure of HCV infection, as the cell susceptibility could be reduced when its interfering RNA was introduced.19 By receptor-mediated endocytosis, the viruses are engulfed to form endosomes. The endosomes are acidified in the cytoplasm afterwards. The decreased pH value in the micro-environment activates the fusion between viral envelope glycoproteins and endosome membranes. Then, the core particle is released into the cytoplasm and core proteins are digested to release the viral RNA subsequently.11
Translation and replication of HCV RNA
The IRSE locating on the 5’-UTR is the foundation of viral protein translation. It consists of 3 stem-loop structures and several codons at the beginning of the ORF. When combined with the 40S subunit of the ribosome, the ribosomal conformational change would be triggered to form an activated 80S ribosome.6 Then the translation of proteins starts.
Like all the viruses with a plus strand RNA genome, the replication of HCV RNA occurs on the endoplasmic reticulum. An HCV replication complex (RC) is formed when the non-structural proteins anchored on the membrane of the endoplasmic reticulum. But the definite components of an HCV RC remain unclear. It can only be identified that the viral genome RNA, non-structural proteins, and cooperative factors are included. The part of endoplasmic reticulum where an HCV RC formed would invaginate to be a cystic structure harboring all the functional RC components. In the cystic cavity, the progeny RNA is synthesized with the viral genome RNA inside HCV RC serving as the template.20,21
The assembly and release of progeny viral particles
Lipoproteins are thought to concern with the maturity of viral particles, playing an important role in the particle assembly and release. In some viral particles, the human apolipoproteins were detected. If the expression of apolipoproteins were down-regulated, the production of progeny viruses would be reduced obviously.22,23 The virus assembly is considered to take place in the intracellular lipid droplets as the HCV core proteins were found on the external surface of lipid droplets in the infected cells.24 Moreover, NS2 protein forms complexes with structural proteins including E1 and E2, as well as p7 and non-structural proteins such as NS3 and NS5A near the lipid droplets. Ma et al’s25 study in 2011 suggested that it is the unique HCV protein that interacts with both structural and non-structural proteins. From their result, they speculated that NS2 formed 2 complexes for viral assembly. One is with E1, E2, and NS3 while the other with NS5A. The former works on initial assembly and the later on post maturation. The p7 mutation would interrupt the interaction between NS2 and other viral proteins to reduce the virus production. Because the absence of normal p7 would induce an endoplasmic reticulum-like distribution of NS2 and the aberrant distribution would lead to less effective viral assembly and maturation. These effects might result from the alteration in the membrane topology of NS2 C-terminal domain induced by p7 mutations. We have already known that RC would be directed to the place adjacent to LDs by NS5A to form progeny infectious viral particles. Interestingly, the complexes formed by E1, E2, NS2, NS3, and NS5A turned out to coexist with RC and LDs in a dot-like structure in their study. It is inferred that NS2 might serve as a scaffold to mediate the interaction between RC, LDs, and other 2 complexes. However, even we thought that the NS5B protein would work on virus assembly, direct evidence remains to be found. After post-assembly maturation, progeny viral particles are released outside via the secretion pathway of lipoproteins.
The pathophysiology after HCV infection
The immune system could eliminate HCV infection during the acute stage of the infection in a few patients. Unfortunately, most patients switched to chronic infection after ineffective virus elimination. The pathological damage of chronic hepatitis C included cytotoxic T cell-induced and oxidative stress-induced liver damages.26 It needs to be emphasized that oxidative stress played an important role in virus-induced liver damage. The virus infection could initiate the formation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) beside mitochondria functional loss. When ROS increased, the transforming growth factor-β (TGF-β) was up-regulated to promote liver fibrosis.27,28 The increase of intracellular ROS/RNS was also a precipitating factor of genetic mutation and DNA damage that would accelerate the occurrence of hepatocellular carcinoma afterwards.29 Moreover, the core proteins were found involved in HCC pathogenesis after HCV infection. By inducing insulin resistance and LDs formation, core proteins cause the hepatocytes to adipose degeneration. And under the condition of liver fibrosis, HCC would easily happen after hepatic steatosis.30,31 In the meantime, HCV proteins including core proteins were confirmed effectively in cell proliferation regulation, which made them potential carcinogenic elements. Core proteins would activate transcription factors and proto-oncogene to inhibit cell apoptosis, provoke expression of growth factors or related receptors and regulate tumor suppressor gene p53. NS3 and NS5A proteins could interact with p53 to inhibit the transcription activation and cell apoptosis as well.32-34 Tumor suppressor gene pRb was another target for HCV viral proteins, interacting with both core protein and NS5B.35 NS5B could directly stimulate the infected cells to activate E3 ubiqutin ligase E6AP, resulting in pRb ubiquitination and secondary degradation.36
As a membrane-associated protein, NS4B has four transmembrane domains consisting of highly hydrophobic peptide. It could regulate NS5A phosphorylation and NS5B RNA-dependent RNA polymerase function as well as adjust the valid location of membrane-associated complexes, RC, and HCV RNA.37 The interaction between NS4B and oncogene Ha-ras is extremely important in HCV related HCC occurrence. When Ha-ras was activated by NS4B, the cell morphology and growth characteristics would alter.38 Moreover, its nucleotide binding motif (NBM) mediated transformation activity would induce aberrant proliferation of hepatocytes in both in vitro and in vivo experiments. After NBM inhibited, the RNA replication and related HCC formation could be inhibited either.39
Direct-acting antivirals targeting on HCV viral proteins
Antiviral therapy could prevent disease progression and provide long-term benefit, if successful, because lifelong sustained virological response (SVR) usually results in improved life quality and survival rate.40 Pegylated interferon plus ribavirin had been the standard of care (SOC) for hepatitis C over a decade. However, the combination regimen only achieved SVR in 40%-50% of genotype 1 HCV infected patients41,42 worldwide despite the IL-28 genotype diversity in different nationalities.43 The SVR might be higher in other genotypes, but still, more than a quarter of patients were non-responders. The SOC combined immunomodulation and antivirus, but antiviral efficacy of ribavirin was weak and nonspecific. Along with further investigations in HCV molecular characteristics, the processes of virus entry, decortication, translation, hydrolysis of precursor poly-proteins and progeny virus assembly may all be considered as targets for more effective antiviral agents. Viral proteins played important roles in these procedures. As the newly emerged agents exactly targeting on some of them to interrupt life cycle of the virus, they showed better efficacy on virus eradication and SVR, shorter treatment duration and less side effects in different regimens compared to SOC. The common therapy is now gradually shifting to an era of direct-acting antivirals (DAAs).44
Current DAAs could be divided into four different groups: N3/4A protease inhibitors (PIs), NS5A protein inhibitors, nucleoside type NS5B polymerase inhibitor (NIs) and non-nucleoside type NS5B polymerase (NNIs).41 Although plenty of DAAs were being developed, only seven DAAs were approved by European Union and recommended in different regimens in the latest update of EASL guidance.44 They were simeprevir and paritaprevir of PIs, daclatasvir, ledipasvir, and ombitasvir of NS5A protein inhibitor, sofosbuvir of NIs and dasabuvir of NNIs45 (Table 1).
Current approved direct-acting antivirals (DAAs) and their targets (based on Euro union approved drugs).
NS3/4A protease inhibitor
PIs worked on the N3/4A protease by influencing hydrolysis of precursor poly-proteins. They were the earliest DAAs as telaprevir and boceprevir were the first 2 approved agents. However, these 2 PIs were only approved in genotype 1 HCV infection and would cause side effects including anemia and rash as strong and moderate cytochrome P450 (CYP) 3A inhibitors respectively.46 They were not first line recommended DAAs any more.44
As a mild inhibitor of CYP1A2 and intestinal CYP3A, simeprevir has fewer side effects.46 It was designed to be used simultaneously with other antiviral drugs to improve treatment efficacy, but coadministration with ledipasvir/sofosbuvir fixed agent is not recommended for the plasma concentration of latter one would be increased by simeprevir.47
Paritaprevir is a macrocyclic noncovalent peptidomimetic acyl-sulfonamide, and co-administered with CYP34A inhibitor ritonavir could dramatically increase the plasma drug concentration and half-life period. Thus, it is commonly used in a fixed dosage with ritonavir. Like first generation PIs, resistance-associated variants (RAV) emerged quickly after monotherapy, usually presenting as amino acid 155 and 168 substitutions in genotype 1. The combination regimen with NS5A inhibitor and NNI is more appropriate.48,49
NS5A protein inhibitor
Although lacking known enzymatic function, NS5A seems to be critical in virus replication, not only in RNA synthesis but also in generating a suitable intracellular environment for the synthesis. It is also important in virus assembly, guaranteeing infective ability.48,50 Because of the highly conserved structure of domain 1 of NS5A, their inhibitors have pan-genotypic efficacy but lower genetic barrier to resistance, making them good candidates in combination regimens.51
Daclastavir is a p-glycoprotein substrate metabolized by CYP3A4 with highly protein bound ability. The advantage of daclatasvir is its compatiblity with other drugs, making it usable in patients with complications. It would not change the immunosuppressant concentrations, and coadministration with opioids does not require dose adjustments via evaluation of pharmacokinetic interactions.52,53 Moreover, it is suitable for patients with renal or hepatic impairment because of acceptable variation of drug concentration in such patients.54,55
Ledipasvir is supposed to inhibit hyperphoshorylation of NS5A, which is important for progeny virus production, showing pan-genotypic antiviral efficacy (though lower in genotype 2a and 3a). Some amino acid substitution might generate resistant virus strain such as Y93H or L31V in genotype 1b. Consequently, a combination regimen with NS5B inhibitor sofosbuvir for higher resistance barrier is needed. And NS5A inhibitors seem to have an additive effect when combined with NS5B inhibitors.47,56
The naphthyridine inhibitor was shown to target on NS5A, and after optimization, ombitasvir came out. It is an N-naphthyridine based inhibitor, developed for co-administration with PI paritaprevir and NNI dasabuvir, with RBV, or not, showing a broad genotypic coverage. Resistance occurs when amino acid positions 28, 30, 31, 58, or 93 mutate in NS5A, but the combination would provide a higher resistance barrier in patients to achieve a higher SVR.48
Nucleoside type NS5B polymerase inhibitor
NIs could terminate RNA chain extension by competitively inhibiting attachment of natural nucleosides. All NIs show pan-genotypic antiviral ability. The resistance mutation of serine to threonine in position 282 of NS5B would reduce virus fitness since mutation in RdRp is intolerant. Therefore, such resistance mutation would not cause virological relapse.57 As the only approved agent in this class, sofosbuvir is a star DAA. It’s the pro-drug of uridine triphosphate, binding to the RNA chain after converted to interrupt elongation. Moreover, it has limited only affinity to host RNA or DNA polymerases and no involvement of CYP3A/4 in its metabolism. All these features result in fewer side effects, less drug-to-drug interaction as well as better tolerance.58 Likewise, Sofosbuvir is not recommended for monotherapy. The combination regimen would reduce occurrence rate of S282T mutation which seems not obligated to viral relapse by now but lack of long-term report on patients’ prognosis.59,60
Non-nucleoside type NS5B polymerase inhibitor
Unlike NIs, NNIs are allosteric inhibitors affecting RdRp via allosteric binding. There are at least four different allosteric sites in NS5B, including site I and site II in the thumb domain, as well as site III and site IV in the palm domain. NNIs altered the topology of polymerase when conjugated with specific site in NS5B. As the polymerase couldn’t transform into the active conformation, the viral RNA replication was interrupted. But, such mechanism result in low resistance barrier and virological breakthrough is common in monotherapy. Dasabuvir worked on Site III in the palm domain.41 Another kind of NNIs, GS-9669 worked on the thumb site II of NS5B. The mutations at NS5B positions 419, 422, 423, 482, 486, and 494 confer variants resistance but reduced fitness. Lack of cross-resistance made such agents also good candidates for combination regimens.61
Hepatitis C virus and host protective immunity. When animals were immunized by the envelope proteins of flavivirus, the host would acquire protective antibodies. As a member in flaviviridae, it could be inferred that the HCV envelope proteins are the major antigens to activate host immune response among these viral proteins. The HCV envelope proteins consist of E1 and E2 (ie. gp35 and gp70) proteins. When it came to bovine diarrhea viruses, whose envelope glycoproteins consisted of 2 proteins of different size similar to HCV, the neutralizing antibodies could only be induced by the large protein. Furthermore, the structure of HIV-I gp120 V3 loop containing the neutralizing antigenic epitope domain is similar to the structure of HCV gp70 HVR1. From all these, it could be speculated there might be neutralizing antigenic epitopes in HVR1 of HCV large envelope protein gp70. The HVR1 is found from 390 to 410 amino acid residues, but the precise number and location of neutralizing antigenic epitopes remains unclear. The results from multiple researchers varied but one consensus had been reached: these epitopes were B cell epitopes and served as targets for host humoral immune response.62 In the classical protective immunity model, antibodies induced by neutralizing antigenic epitopes did not only rupture the attachment between viruses and susceptible cells, but also enhance the ability of immune cells for virus identification and elimination. However, when researchers investigated further in HCV, they found a different pattern. In vitro experiments, high potent antibodies for E2 could agglutinate the complexes, which consisted of E1 and E2 non-covalently and located on the surface of HCV particles. Afterwards the combination between E2 and receptors on targeted cells would be interrupted. It seems that the antibodies induced by E2 presented their ability as neutralizing antibodies, but they can only interact with latest parental viruses. Such neutralization was not found in progeny viral particles. One possible explanation might be that, as a rapidly volatile domain, the gene of E2 HVR1 would easily drift under the host immune pressure. It explained that the antibodies induced by parental viruses could only be neutralized with latest viral strains but the progeny mutant strains.63
The antigenic epitopes include linear epitopes and spatial epitopes. From the phage random peptide library, some mimic epitopes with HCV specific antibodies could be screened out. These mimic epitopes differed with original antigens in primary structures but shared the same ability to combine with specific antibodies. Most of them are spatial epitopes. It must be emphasized that mimic epitopes are high antigenic and broad-spectrum. The neutralizing antibodies induced by them could interact with multiple kinds of HVR1 crossways.64 The mimic epitopes might be an effective solution to overcome the polytrope of HCV HVR1 to invent protein vaccine or genetic vaccine.
In conclusion, it could be learned that HCV has unique viral structure and RNA genome. Encoded structural and non-structural viral proteins are essential to its life cycle and pathogenesis. Currently, approved DAAs target on some of these proteins to influence viral life cycle. However, protective immunity is difficult to acquire because of volatility in HCV antigenic epitopes. During the life cycle of HCV, there are many other concerned factors no matter in the parental virus infection link or progeny virus production link. These factors play important roles in interactions among viruses, host cell micro-environment and host immune system.
However, because of limited natural host, the knowledge we learned about HCV characteristics mostly came from vitro studies on subgenomic replicons, pseudovirions, mutant viral particles, and so on. New HCV replication models have been established.65 Such models would open up new ways for researches in viruses infection, replication, and release on the absent of host cell adaptive mutation in virus strain. More information on HCV molecular characteristics and related pathways would be penetrated in a few days. Along with further investigations in HCV molecular characteristics, the developments in direct-acting antivirals might promote better management for patients and the developments in host protective immunity against HCV might prevent susceptible populations from infection.
Copyright
Whenever a manuscript contains material (tables, figures, etc.) which is protected by copyright (previously published), it is the obligation of the author to obtain written permission from the holder of the copyright (usually the publisher) to reproduce the material in Saudi Medical Journal. This also applies if the material is the authors own work. Please submit copies of the material from the source in which it was first published.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
Related Articles
Cited By...
- No citing articles found.