


Abstract
Objectives: To determine the prevalence of Fabry disease (FD) among Saudi patients on hemodialysis.
Methods: This prospective study was conducted in 3 major hospitals in the. All adult patients (>18 years old) attending the dialysis unit who have end-stage renal disease (ESRD) and on hemodialysis were included. Known patients with FD and those who refused to participate in the study were excluded. All eligible patients were screened for FD using dry blood spot (DBS) for alpha-galactosidase A (α-Gal A). A positive DBS (enzyme activity <40%) was followed by another confirmatory enzyme assay. When the second DBS sample was also positive (enzyme activity <40%), a Sanger sequencing of the GLA gene was performed.
Results: A total of 619 patients with ESRD and on hemodialysis were screened for FD using DBS for α-Gal A enzyme level. Enzymatic activity was below 40% in 11 samples. On retesting, 3 females had <20% enzymatic activity suggesting FD. Sanger sequencing of these 3 females showed the variant c.1055C>G (p.Ala352Gly) confirming the diagnosis of FD. Family screening of one of these 3 patients revealed one asymptomatic female carrying the same variant.
Conclusion: The prevalence of FD in this cohort was 4.8 per 1000 patients. Screening of Fabry patients with ESRD seems to be a cost-effective strategy. Furthermore, relatives of the patients identified by screening enhances this screening strategy.
Fabry disease (FD) is an X-linked lysosome-storage disease caused by a deficiency of the enzyme alpha-galactosidase A (α-Gal A), encoded by the GLA gene.1
The genetic basis of FD is well established. According to the human gene mutation database, more than one-thousand mutations in the GLA gene have been described.2 As a x-linked disease, the prevalence of FD in females is predicted to be double that in males.1 There are multiple types of FD-causing mutations.3 The most common are point mutations, including missense or nonsense nucleotide substitution (approximately 70%).2,3 Other variants, such as small deletions, insertions, and duplications, were reported in FD.2,4
Deficiency of α-Gal A activity results in the accumulation of glycosphingolipids (GSLs), specifically globo-tria-osyl-ceramide (Gb3) and globo-tria-osyl-sphingosine (lyso Gb3).5-8 Lysosomal globo-tria-osyl-sphingosine accumulates exclusively in the lysosomes of various body tissues and organs, leading to irreversible cell damage.5,7 Lysosomal globo-tria-osyl-sphingosine has a role in vascular remodeling; it stimulates smooth muscle proliferation and has a role in enhancing the proliferation of cardiomyocyte and glomerular injury.5,7
There is a wide variability in the clinical phenotype of FD. This is observed in the age of onset, the disease progression, and the organs affected.9,10 Furthermore, there is no clear genotype/phenotype correlation.1,9,10 Many genetic and environmental factors were proposed to explain this heterogeneity.1 Nevertheless, most of the patients with FD present with chronic kidney disease, premature stroke, or left ventricular failure.9-12 Heterozygous females tend to be asymptomatic or present with milder symptoms, whereas hemizygous males typically have severe symptoms.1,9,11 The clinical heterogeneity of FD and the nonspecific symptoms make the diagnosis challenging. Moreover, approximately 30% of females with FD have a normal α-Gal A level.1,6,8 Therefore, genetic analysis is required to confirm the diagnosis.
As the clinical diagnosis of FD is challenging; therefore, screening of subjects with clinical suspicion of the disease is important.13-18 Early detection and initiation of enzyme replacement therapy (ERT) may slow the progression of the disease.19-22
Wide-scale newborn screening has been implemented for FD in many countries.1 This allows the early detection and treatment of patients with FD. Screening is based on measurement of alpha-galactosidase A enzyme activity in dry blood spot (DBS) using either tandem mass spectrometry (MS/MS) or fluorimetry.23 This method has also been used for screening of FD in adults with high risk of the disease. If the initial screening test showed low enzymatic activity, another DBS sample is tested to confirm the result.23 Enzymatic assay of lysosomal enzymes including alpha-galactosidase A in DBS has been developed, evaluated and improved in the last 2 decades.24 Previous studies have shown that DBS enzyme assay using MSMS is comparable to the fluorometric method.1,25,26 Moreover, DBS has the practical advantage in universal screening.
Currently, there are no data on the prevalence of FD in the Kingdom of Saudi Arabia (KSA). Here, we screen individuals with a high risk of FD, specifically those receiving hemodialysis who may benefit from ERT. We expect that the data generated in this study will provide insights into the prevalence of FD in KSA. Also, the segregation study of the relatives of the index individual picked by screening helps in early diagnosis and initiation of ERT for asymptomatic subjects. Therefore, this study aimed to determine the prevalence of FD among adults on hemodialysis who attend 3 tertiary centers in KSA.
Methods
This prospective study enrolled adult patients (>18 years old) with unexplained end-stage renal disease (ESRD) and on maintenance hemodialysis. The study was conducted in 3 referral hospitals in KSA. which included 2 hospitals in the central region: Prince Sultan Military Medical City (PSMMC), Riyadh, KSA (with more than 1500 beds; 450 beds in the dialysis unit), King Khalid hospital in Al-Kharj (with 385 beds), KSA, and Tabuk Military Hospital, Tabuk, KSA, which is located in the northern region (with 350 beds). Patients were recruited over one year between January 2018 and January 2019. All adult patients (>18 years old) with unexplained ESRD on hemodialysis attending these 3 hospitals were included. Known patients with FD and those who refused to participate in the study were excluded. Also, patients with a known cause of ESRD, such as obstructive uropathy, were excluded. The diagnosis of ESRD was established by the standard biochemical, radiological, and histological tests.1 Data was abstracted from the laboratory and clinical records using case report forms.
The diagnosis of FD in all enrolled patients was confirmed by enzyme assay analysis (alpha-gal-A enzyme activity in white blood cells) and DNA sequence analysis.
Informed consent was offered to the patients before enrolment in the study. Patients who refused to participate in the study were excluded. The institutional research board at PSMMC approved this study.
Screening and diagnosis of Fabry disease
Screening for FD was performed by measuring α-Gal A activity in DBS. Blood was spotted directly onto the filter paper using a Lancet finger stick. Blood spots were dried for at least 4 hours at room temperature and stored in sealed plastic bags at 4°C. Then samples transported to the laboratory for measurement of the enzyme activity using a fluorimetric method.8 A positive DBS (enzyme activity <40%) was followed by another confirmatory enzyme assay. When the second DBS sample was positive (enzyme activity <40%), molecular confirmation is requested. Three ml of Ethylenediaminetetraacetic acid blood was taken for deoxyribonucleic acid extraction and amplification by Polymerase chain reaction, followed by Sanger sequencing of the GLA gene.3 The diagnosis of FD was considered confirmed if a variant is detected in the GLA gene. Given the higher probability of FD and the fact that enzymatic activity can be normal in Fabry females, all first-degree relatives of the confirmed cases were offered both enzymatic and genetic testing to screen for the disease.
Statistical analysis
The statistical analysis was performed using the Statistical Package for Social Sciences software, version 22.0 (IBM Corp, Armonk, NY, USA). Categorical variables were presented as frequencies and percentages, while continuous variables were presented as mean and standard deviation. A p<0.05 was considered significant.
Results
A total of 619 patients with ESRD and on hemodialysis were screened for FD using DBS for α-Gal A enzyme level. Enzymatic activity was below 40% of the normal level in 11 samples. On retesting of these 11 subjects, 3 females were shown to have <20% enzymatic activity suggesting FD (Table 1). Sanger sequencing of these 3 females showed the variant c.1055C>G (p.Ala352Gly), confirming the diagnosis of FD. Therefore, the prevalence of FD in this cohort of patients with ESRD and on hemodialysis was 4.8 per 1000 patients.
Characteristics of patients with end-stage renal disease who underwent screening for Fabry disease.
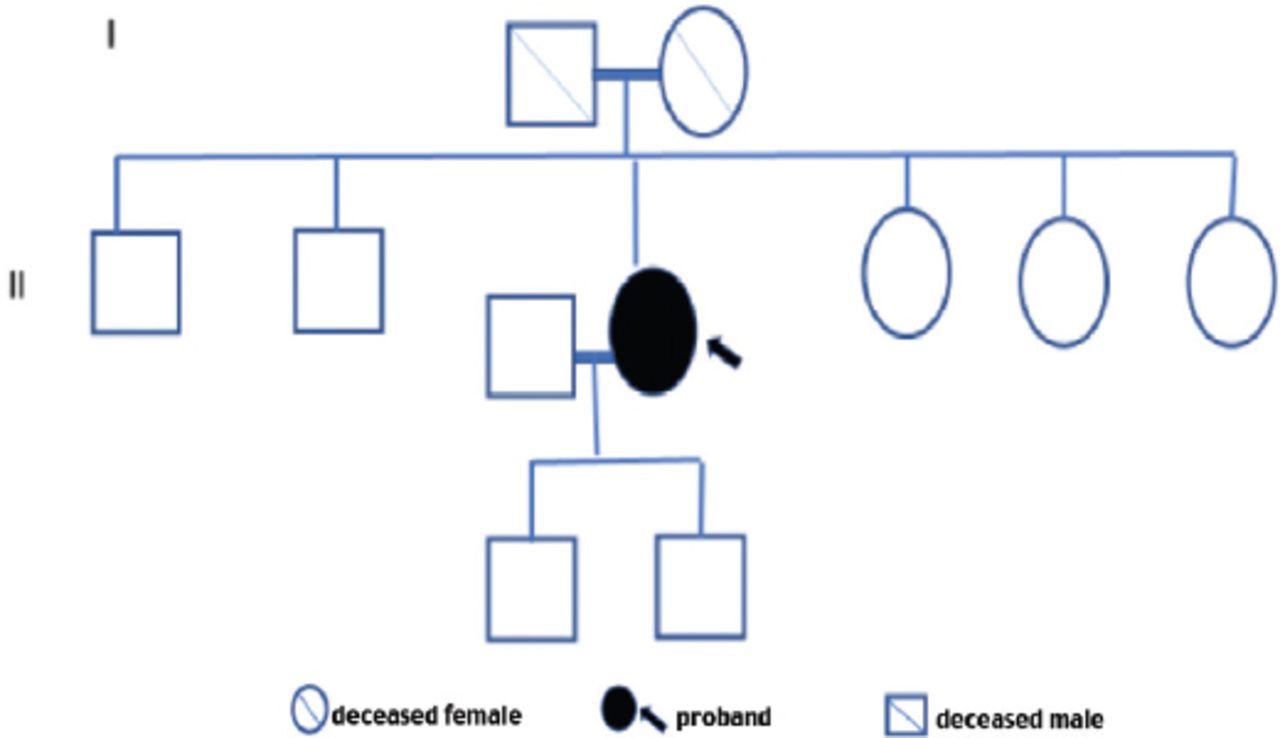
Family screening of one of these 3 patients revealed one asymptomatic female sibling carrying the same variant. All first-degree relatives of the other 2 patients with FD failed to consent for screening, and therefore these persons were not tested. The 3 confirmed patients had no family history of kidney disease or FD (Table 2). The family pedigree of the 3 patients is shown in Figures 1-3. A detailed history and clinical examination failed to show any symptoms or signs related to FD except for chronic kidney disease (Table 2). Specifically, there were no skin, eye, or cardiovascular manifestations. Also, electrocardiogram and echocardiography were normal. Renal biopsy was not performed in any of the 3 patients because the kidney is expected to be small and fibrotic, and therefore, the findings are unlikely to be informative.
Characteristics of patients with a confirmed diagnosis of Fabry disease.
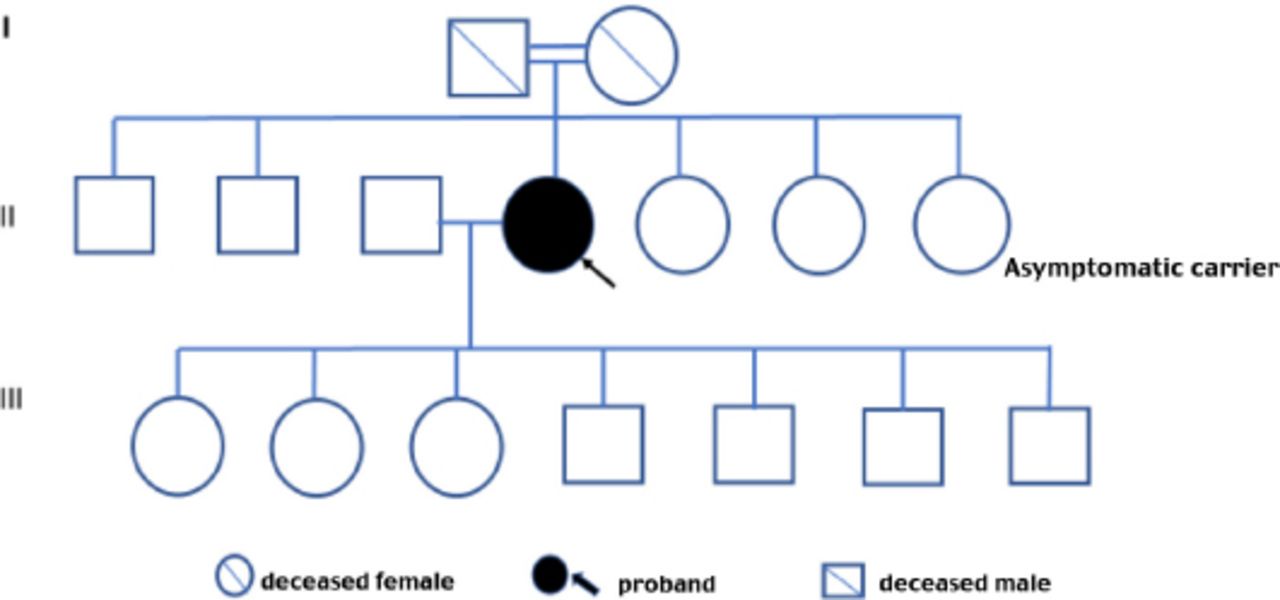
Family pedigree of patient 1.
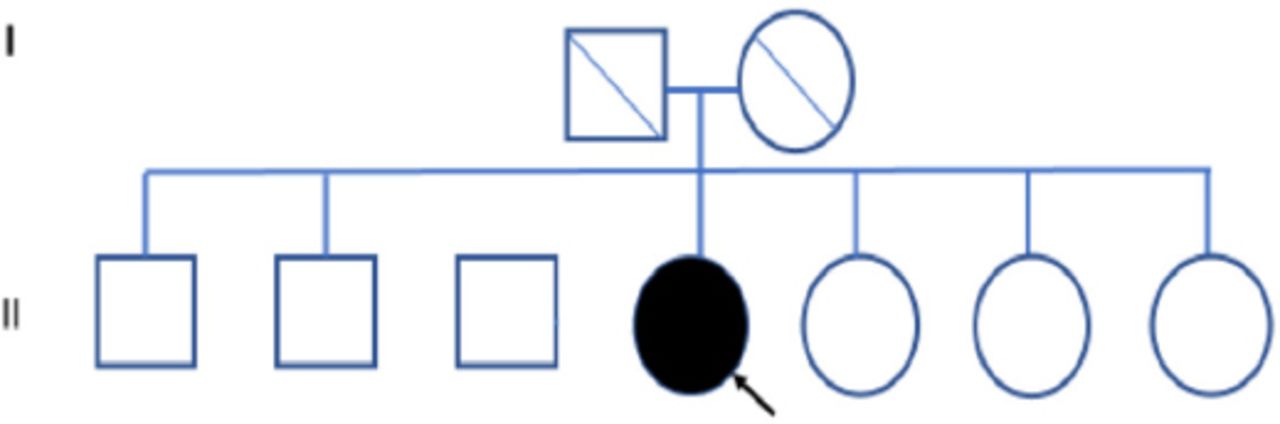
Family pedigree of patient 2.

Family pedigree of patient 3.
Discussion
Fabry disease is a progressive disorder that affects multiple organs, including the kidney, the heart, and the brain. Its diagnosis is challenging because the symptoms and signs of the disease are nonspecific and, therefore, can be overlooked. It seems that FD is underdiagnosed, especially in Arab countries. This is evident from the few reported cases from the region. Also, there are few Arab patients with FD included in the Fabry registry.11 This registry was initiated in 2001 and considered as the largest observational database in the world for orphan diseases; most of its recruited patients were from Europe and North America.11 Universal newborn screening might be the only reliable method to ascertain the epidemiological data on FD and to optimize its management by early detection and starting ERT.22 Nevertheless, screening of high-risk populations might be more practical and could identify patients with late-onset manifestation.13 This encouraged us to screen for FD among our patients with ESRD.
The prevalence of FD in our cohort of 619 Saudi patients with ESRD and on hemodialysis was 4.8 per 1000 patients. A similar approach was adopted in previous studies, which reported a prevalence of 0.2-1.7% of FD in patients with chronic kidney disease depending on the population screened and the method of screening.1,10-18 Linthorst et al,13 conducted a systematic review of FD screening of high-risk populations. The authors showed that the average prevalence of FD among hemodialysis patients was 0.33% in males and 0.10% in females. Our cohort included both males and females and showed a higher prevalence of FD among hemodialysis patients. This indicates that FD is underdiagnosed in our institutes. Similarly, Sayilar et al,16 screened 1527 patients on hemodialysis (n=1435) or peritoneal dialysis (n=92) for FD across 17 dialysis centers in Bursa province, Turkey. The authors reported a 0.3% overall prevalence of FD among Turkish patients.16 Similarly, a Russian nationwide FD screening among 5572 dialysis patients revealed a prevalence of 0.36%.18 In contrary to our results, a recent study from Thailand failed to detect any definitive FD patients in a cohort with ESRD of unknown etiology.17 This could partially be explained by the small population screened (124 patients). This suggests that it is necessary to include larger centers in screening trials of orphan diseases to increase their statistical power.13
In our cohort, 3 females showed enzyme activity of <20% after retesting. They all carry the variant c.1055C>G (p.Ala352Gly). This variant causes an amino acid change from Ala to Gly at position 352. According to study in 2018, this variant has previously been described for Fabry disease by Lukas et al,2 who showed that the variant leads to an enzymatic activity range of 53.7±4.2 relative to wild-type activity in vitro.3 Furthermore, in vitro studies showed that this variant is deoxygalactonojirimycin responsive.27,28 This drug is used for treatment of FD.1 Similarly, this variant was reported as likely pathogenic by Bioscientia institut fuer medizinische diagnostik GmbH, Sonic Healthcare in Clinvar database.27 Other clinical, biochemical and computational evidence suggesting pathogenicity of this variant include: i) The clinical phenotype of the 3 patients is consistent with FD, as all of them presented with unexplained ESRD, which is a known feature of the disease; ii) The presence of a single variant in 3 unrelated patients with a phenotype consistent with FD suggests that this mutation is likely to be disease-causing; iii) The α-Gal A enzyme was low in the 3 patients on 2 occasions; iv) In Silico prediction studies indicate that this variant is likely damaging and disease causing; v) This variant has a low allele frequency in the Western population.3,27 However, this variant was reported in 10 patients in Saudi Genome Project that lists a database of over 6300 variants in different monogenic disorders.29 Therefore, further functional and population studies of the variant we identified is needed to ascertain and replicate our findings and to give more insight into the genotype phenotype correlation of FD in the Arab region and to compare it with the rest of the world. Nevertheless, We have shown evidence from the clinical presentation (unexplained ESRD), low enzyme activity, functional studies and in Silico prediction studies that the variant we detected is likely pathogenic.
We did family segregation study for patient one. The other 2 families declined to consent for segregation. This study showed a 20-year-old female sibling of patient one is a carrier of the same variant in a heterozygote status. Her α-Gal A enzyme was <20%. However, her renal function was intact. She was put under surveillance and close monitoring.
Lysosomal globo-tria-osyl-sphingosine was normal in our 3 patients with FD. This biomarker is known to correlate with the severity of the disease, and it has been shown to be high in male patients and normal or mildly elevated in females, which was the case in our 3 female patients.7,8
Study limitations
The main limitation of this study is that we relied on the α-Gal A enzyme level as the only screening tool. It is well known that females and also some mild pathogenic variants in males have residual enzyme activity. Thus, some FD patients might have been missed. Adding other biomarkers to the screening strategy, such as Lyso Gb3, could have improved the accuracy of our case finding.7 Also, we were unable to complete the family segregation for logistic reasons and family denial. Family screening could have detected more cases.
In conclusion, FD is common in Saudi patients with ESRD on hemodialysis. Therefore, it seems that FD is more common in KSA than previously thought. This study indicates that screening of patients with ESRD for FD seems to be a cost-effective case-finding strategy. Furthermore, screening of relatives of patients detected by screening might further enhance this screening strategy.
Acknowledgment
The authors gratefully acknowledge all the patients and families who participated in this study. The authors would also like to thank Prince Abdullah bin Khalid Celiac Disease Research Chair, Vice Deanship of Scientific Research Chairs, King Saud University, Riyadh, Kingdom of Saudi Arabia, for the support. We also like to thank Sanofi company for assisting with the enzyme assay and molecular testing for the patients. We would also like to thank the American journal expert for English language editing.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received April 6, 2020.
- Accepted June 24, 2020.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.