


Abstract
Objectives: To identify the novel and promising indicators for pulmonary tuberculosis (PTB) patients
Methods: The study was carried out between June 2016 and June 2019. Three RNA sequencing or microarray datasets of TB infection were used to identify the potential genes showing a common expression trend. The expression level of screened targets was determined by reverse transcription polymerase chain reaction and ELISA using samples of whole blood and peripheral blood mononuclear cells (PBMCs) isolated from 69 PTB patients and 69 healthy volunteers. The potential of the identified targets to predict the treatment outcomes was further studied.
Results: Bioinformatics analysis demonstrated that a total of 91 genes were up-regulated in all the 3 datasets; among them, the expression of SLAMF8, LILRB4, and IL-10Ra was significantly increased at both the mRNA and protein levels in whole blood and PBMC samples of PTB patients compared with the healthy controls. The mortality rate increased significantly in SLAMF8 or LILRB4 high expression group compared with SLAMF8 or LILRB4 low expression group. Further, the decrease rate of bacteria in patients with SLAMF8 or LILRB4 high expression was slower than that in patients with SLAMF8 or LILRB4 low expression.
Conclusion: This study provides a promising way to identify novel indicators for PTB. Moreover, the LILRB4 expression may play a role in predicting the outcome of treatments on PTB patients.
Pulmonary tuberculosis (PTB) is caused by the bacterium Mycobacterium tuberculosis (M. tuberculosis). It is the second leading infectious cause of death worldwide.1 According to the World Health Survey, approximately 1,000,400,000 new cases of PTB are reported and approximately 1,800,000 people die each year due to tuberculosis infections. Out of the newly reported cases, approximately 3 million people lack the proper treatment as the current diagnostic methods are inefficient to detect PTB at an early stage due to the emergence of drug-resistant strains of M. tuberculosis.2
Identification of novel biomarkers at an early stage is important for the proper diagnosis, prognosis, and treatment of PTB. Sputum mycobacterial culture is the gold standard for diagnosis and prognosis of PTB, however, the method is slow which takes approximately 1 to 3 months for detection. Acid fast bacilli (AFB) staining using the sputum sample is another common method for PTB diagnosis, however, the method has a limitation of low detection rate.3 In view of these limitations, identification of novel biomarkers is necessary to carry out rapid and efficient clinical practices for PTB.
Progression to active PTB is almost universally regarded as a ‘failure’ of the host immune system to control the infection.4 The process is usually accompanied by the molecular dysregulation of the critical balance between the host cell and M. tuberculosis, which can be considered as a potential predictor for the outcome of infection.5 Reasonably, induced proteins, which were negatively related to the immune response, were considered as the factors of tuberculosis immune escape from the macrophage resulting in poor outcomes.
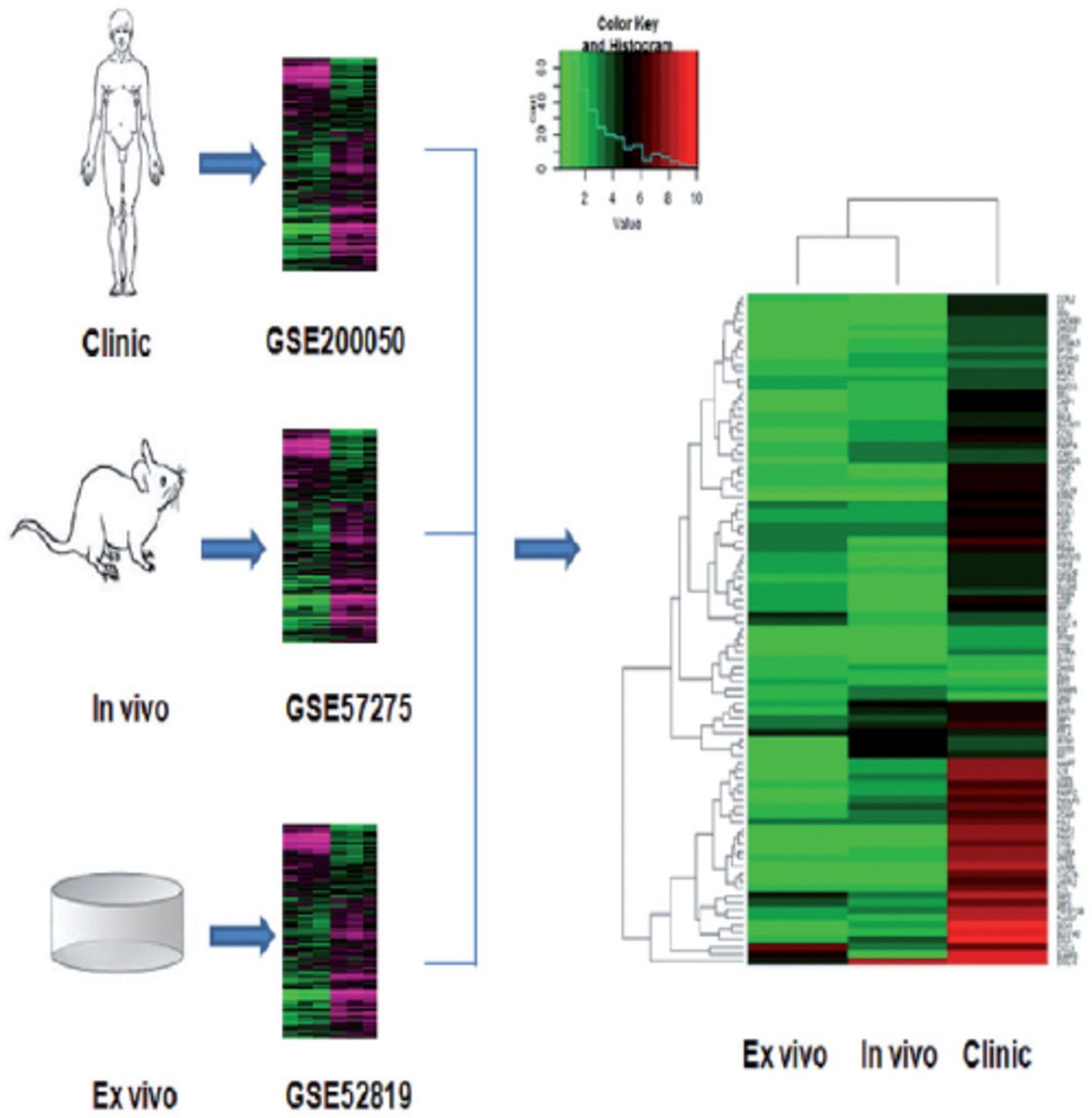
In order to understand the molecular mechanisms underlying the pivotal M. tuberculosis-host interactions, the techniques of ribonucleic acid (RNA) sequencing and microarray have been widely used to analyze the changes in global mRNA expression profiles of cells infected by M. tuberculosis. The cells were obtained for analysis from clinical patients, mouse models of M. tuberculosis infection and cultured cells infected with M. tuberculosis. Various genomic data sets are available online which can be used to compare and analyze the changes in gene expression between the infected and control groups. For instance, Kim et al6 examined the global mRNA expression profiles of caseous human PTB granulomas macrophage (GEO accession number GSE20050, clinical model), Singhal et al7 analyzed the gene expression changes in the lung cells of M. tuberculosis infected mouse (GEO Aaccession no. GSE57275, in vivo model) and Mark et al8 examined the gene expression data from H37Rv infected THP-1 macrophage cells (GEO accession number GSE52819, cultured cells based model). The harmonized datasets obtained from either in vivo or an in vitro models of infection are lacking which can be useful for identification of discriminatory biomarkers of risk in the peripheral blood response of PTB patients. The complete mRNA expression profiles obtained from different models have been observed to vary significantly due to different stages of tuberculosis development. The information on the common gene expression patterns of the different tuberculosis models is available to a lesser extent. Therefore, we proposed to identify and explore these common features in our study which can help elucidate the nosogenesis of M. tuberculosis infection.
The present study reports the characterization of potential PTB biomarkers by exploring the common features of the whole mRNA expression profiles obtained from different PTB models. Studies involving screening and validation experiments have identified various proteins including SLAMF8, LILRB4 and IL-10Ra which were negatively related to immune response and their level was significantly increased in PTB patients as compared to healthy control group. It opens up the possibility that the proteins LILRB4 and SLAMF8 may be used as potential diagnostic biomarkers for PTB development and treatment outcome. As blood sample is easily available for clinical laboratory experiments, and our newly identified protein molecules are related to the specific mechanism of immune escape of tuberculosis, the outcome of the present study may assist in rapid and sensitive detection of this disease. This will further help overcome the pitfalls of existing diagnostic tools.
Methods
We worked on this prospective clinical study in People’s Liberation Army (PLA), Beijing, China 309 hospital (the hospital has been renamed the Eighth Medical Center, Chinese PLA General Hospital) between June 2016 and June 2019.
We used PubMed modular of National Center for Biotechnology Information (NCBI) website to search for prior related publications. We used Datasets modular of NCBI website to search for prior related datasets.
The gene expression profile data was normalized using the IntClus package in R (3.5.1) by the quantile method and filtering out genes with missing values greater than 75%. The summary statistics (Beta, SE, z-value, p-value, I2, H2) was obtained for each gene in all datasets. Advanced methods were implemented for testing model coefficients and confidence intervals using the Metafor package. Following normalization, the integrated gene expression profiles of significantly differentially expressed genes (DEGs) for each dataset were clustered and plotted as heatmap for visualization using the Pheatmap and RColorBrewer package in R (3.5.1). Furthermore, the intersection of the DEGs among the datasets was calculated, and the result was visualized as a Venn diagram using an online tool. The cut-off values used to designate up and down regulation is more than 1.5 fold change and p<0.05. Arbitrary fold change (FC) cut-off of >1.5 and significant p-value of <0.05 was applied to obtain the up- and down-regulated genes.
A total of 69 PTB subjects were included in this study. The inclusion criteria of PTB subjects was described as following: i) refer to the gold standard of PTB diagnosis, subjects whose sputum culture was positive; ii) subjects whose sputum smear was positive, and imaging diagnosis also indicated PTB; or iii) subjects who have granulomatous histopathology of transbronchial lung biopsy and consistent clinicoradiological findings. Patients younger than 18 years old or with incomplete admission or follow-up data were excluded from this study. A total of 44 chronic obstructive pulmonary disease (COPD) patients and 69 healthy volunteers, who were age and gender matched with the PTB subjects, were collected as controls for this study. Tuberculin skin test (TST) were performed for every healthy volunteers and the cases with TST positive were excluded. All patients received the following anti tuberculosis treatment: isoniazid (Anyang First Pharmaceutical Factory, Anyang, China), 300mg/day; rifampin (Suzhou pharmaceutical factory, Suzhou, China), 600mg/day; pyrazinamide (Chengdu first pharmaceutical factory, Chengdu, China), 1.6g/day; and ethambutol (Baiyunshan Pharmaceutical General Factory, Guangzhou, China), 1.2g/day, under the directly observed therapy strategy, as proposed by the World Health Organization. The patients were followed up, and were divided into SLAMF8 high or low group, LILRB4 high or low group and IL-10Ra high or low group with the median expressions level of SLAMF8, LILRB4, or IL-10Ra to compare their treatment outcome after treatment completion based on the median expressions level of certain targets as a cutoff value.
All PTB patients were drawn once a week from day 0 after taking antituberculosis drugs. The blood samples of patients were stored at 4°C for post analysis. Patient sputum samples were collected to monitor the acid-fast smear results and the amount of colony forming units (CFU) every week.
The acid-fast smear staining method is described as follows: hold the smear specimen with a slide, add 2 to 3 drops of carbolic acid and fuchsin, slowly heat the specimen at the height of the flame for 3 to 5 minutes (min), wash the specimen with water after cooling, decolorize it with 3% hydrochloric acid alcohol for 30 seconds to a minute; then wash it with water again, re dye it with alkaline methylene blue solution for one minute, wash it with water, dry it with absorbent paper, and observe it with an oil microscope.
Colony forming units determination was performed by adding 4 fold volume of 4% sodium hydroxide solution to the sputum sample, and let it stand for 30 minutes until it is fully liquefied. The liquefied sputum samples were diluted 2-fold on Middlebrook 7H10 agar media and the CFU were counted 28 days after inoculation.
Total ribonucleic acid extraction and quantitative RT-PCR
Peripheral blood mononuclear cells (PBMCs) were isolated from the whole blood of PTB patients and healthy control group by density-gradient centrifugation method using Ficoll medium (Amersham Pharmacy Biotechnology, Uppsala, Sweden). Total RNA was extracted from the whole blood and PBMC by TRIzol method. The TRIzol reagent was added in 10 fold volume and mixed with 250 µL of the whole blood and then incubated for 5 minutes at room temperature (RT). In case of PBMCs, 107 cells were taken, mixed with one mL of TRIzol and incubated for 5 minutes at RT. Two hundred µL of chloroform was added to each sample, incubated for 3 minutes at RT, and then centrifuged at 12000 g at 4°C. The upper liquid phase was transferred to a 1.5 mL eppendorf tube, mixed with 500 µL of isopropanol by inverting up and down, incubated for 10 minutes, and then centrifuged at 4°C at 12000 g for 10 min. The supernatant was discarded, one mL of 75% ethanol was added to the pellet and mixed by inverting up and down. Again the centrifugation was carried out and the supernatant was discarded. The pellet was dried at RT and 50 µL of RNase free water was added to dissolve the RNA.
The cDNA was synthesized from isolated RNA using the cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, USA) by using iQ5 PCR system (Bio-Rad, Hercules, CA, USA). The conditions used for reverse transcription were as follows: 65°C for 5 min, 37°C for 15 min. followed by 98°C for 5 min. The standard protocol was followed and amplification was carried out by using the following primers: Primer sequences for SLAMF8 was forward: AGCCCTACTTCCCATTACAGT; reverse: AGAGATCGCCAGATAGCCTCA. Primer sequences for LILRB4 was: forward: CATCCATGACAGAGGACTAT; reverse: GGGCTGAAAGGGTGGGTTT. Primer sequences for IL-10Ra was forward: CCTCCGTCTGTGTGGTTTGA; reverse: CACTGCGGTAAGGTCATAGG. Primer sequences for CLEC7A was: forward: CAACACCTCCCTACAAGGAC; reverse: CAGACCTCTGATTGAGCCA. Primer sequences for RELB was: forward: CCATTGAGCGGAAGATTCAA; reverse: CTGCTGGTCCCGATATGAGG. Primer sequences for β-actin was: forward: TCCTCTCCCAAGTCCACACAGG; reverse: GGGCACGAAGGCTCATCATTC. The thermal cycling conditions for PCR were: 95°C for 30 seconds, 95°C for 5 seconds and 40 cycles of 5 seconds at 60°C followed by 72°C for 30 seconds.
Peripheral blood mononuclear cells protein extraction and ELISA assay
Peripheral blood mononuclear cells samples were then suspended in ice-cold lysis buffer (50 mM Tris [pH 7.4], 5 mM Ethylene Diamine Tetraacetic Acid [EDTA], 250 mM NaCl, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 5 µg/ml aprotinin, 5 µg/ml leupeptin, and 1 mM sodium orthovanadate), homogenized, and incubated in ice for 20 min. Samples were centrifuged at 14,000 r.p.m. for 10 min, and the supernatants were collected, quantified by Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA). The targets levels of these samples were determined using commercially enzyme-linked immunosorbent assays (ELISA) according to the manufacturer’s protocol. Enzyme-linked immunosorbent assays kits for SLAMF8 (accession no.: Q9P0V8), LILRB4 ( accession number: Q8NHJ6), and IL-10Ra (accession number: Q13651) were purchased from RayBiotech (Minneapolis, MN, USA) .
Means ± SD was used to express the results from the conducted case versus control study. Student’s t-test was used to determine the differences between 2 groups, and the comparison of the survival time between groups were determined using Kaplan-Meier analysis. All the statistical analysis were performed using Statistical Package for Social Sciences for Windows, version 19 (IBM Corp, Armonk, NY, USA). The difference significance was defined as p-value <0.05.
Results
Common gene expression analyses of GSE20050 (clinic), GSE57275 (in vivo) and GSE52819 (cultured cells based) datasets
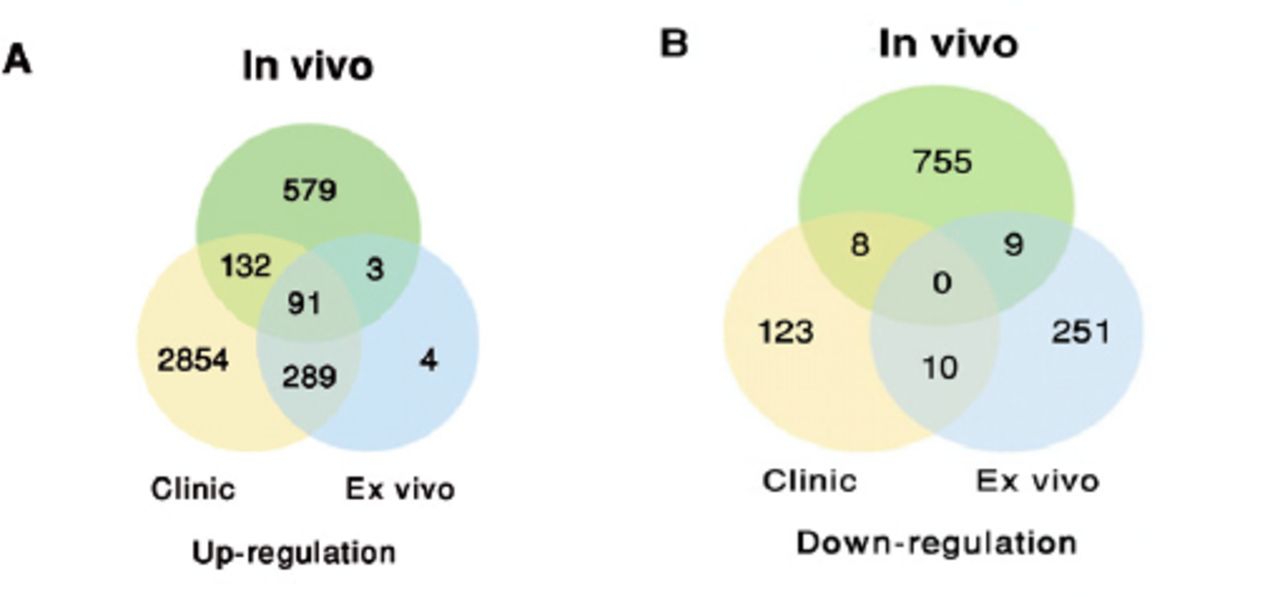
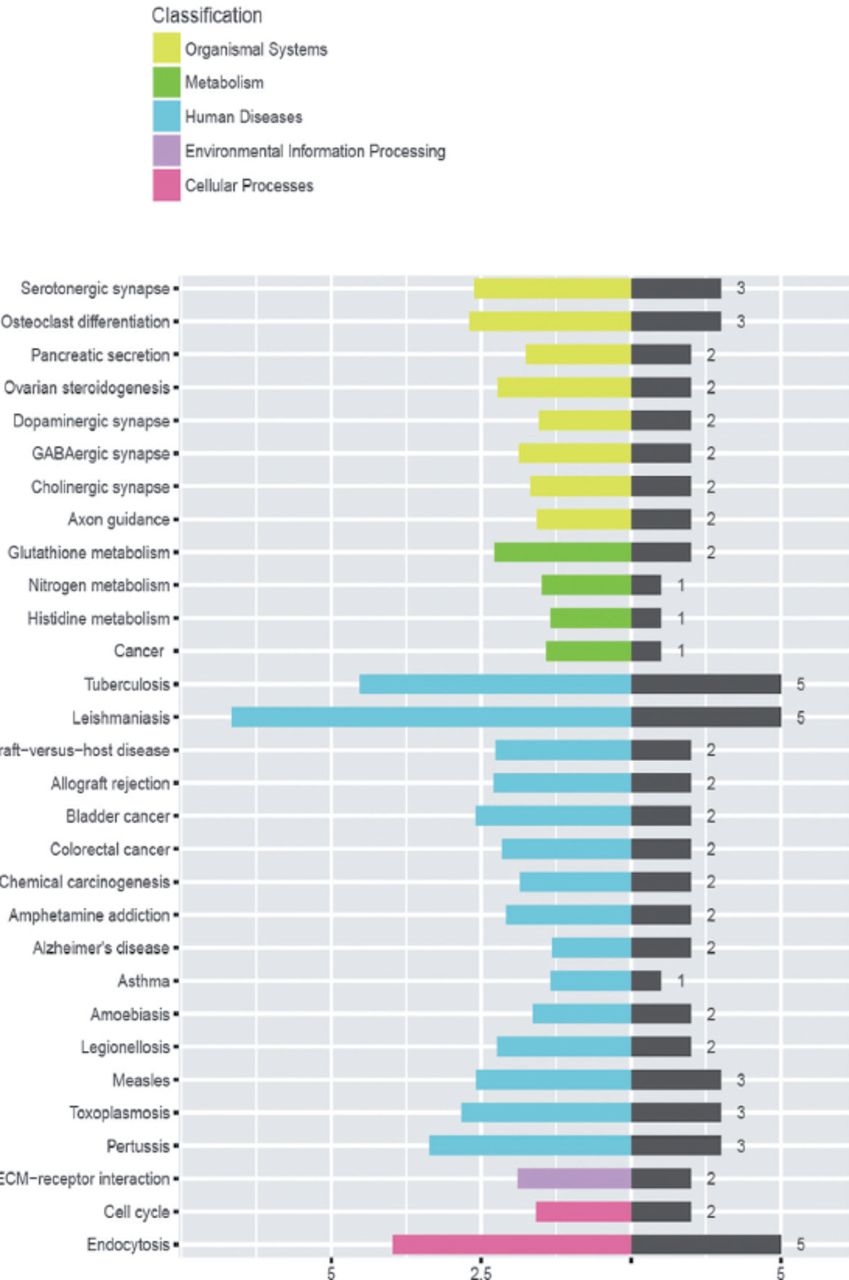
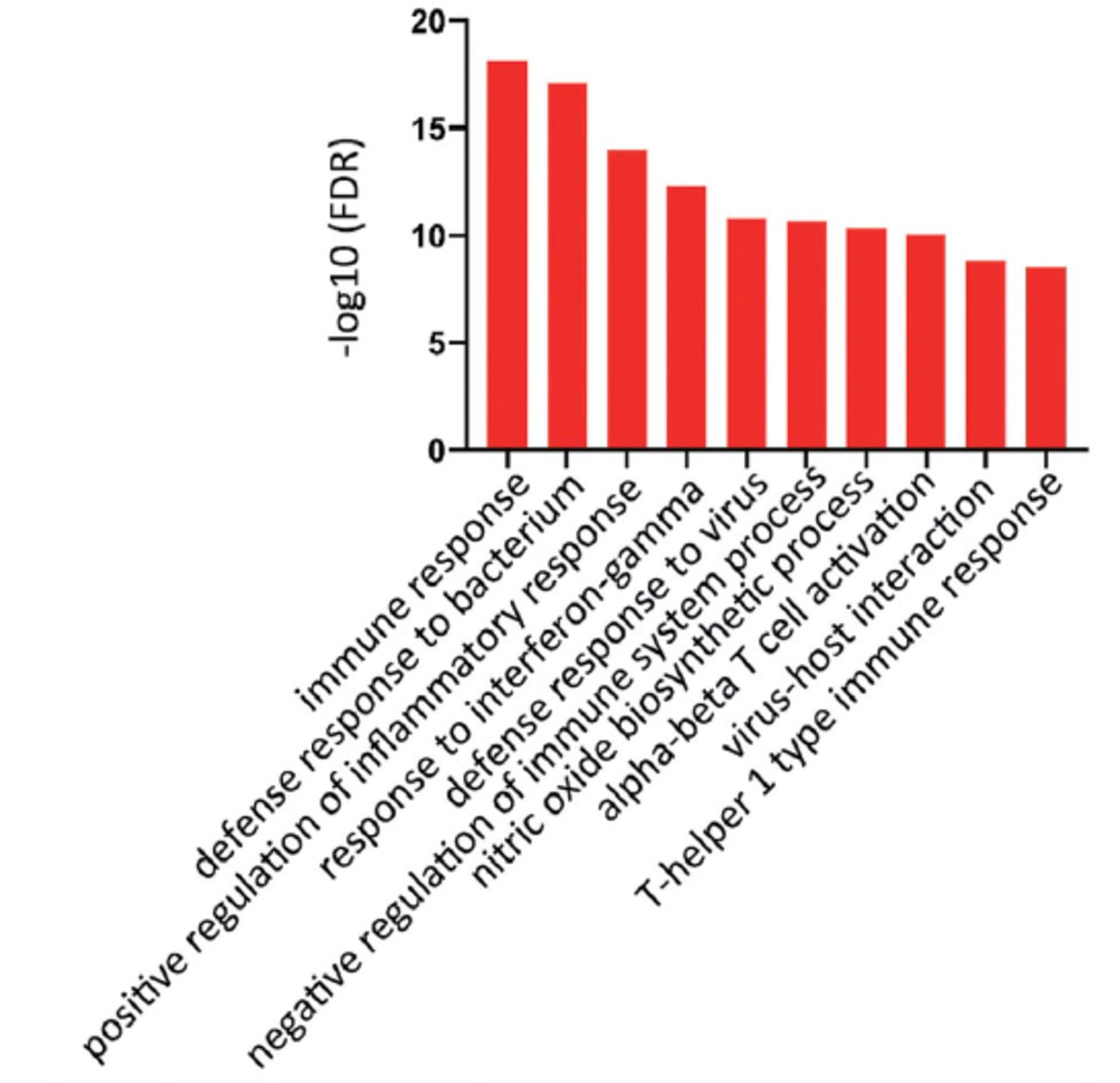
GSE20050 (clinic), GSE57275 (in vivo) and GSE52819 (cultured cells based) datasets were used to identify the common gene dysregulation during M. tuberculosis infection by comparing with the healthy control groups (Figure 1). Venn diagram showed that there was an increase of greater than 2-folds in the expression of 91 targets and no targets were down-regulated in all the 3 datasets (Figure 2). Kyoto Encyclopedia of Genes and Genomes enrichment analysis demonstrated that many of these targets were associated with M. tuberculosis infection. Gene ontology biology process analysis revealed that a small set of proteins (5 targets) were negatively related to immune response including SLAMF8, LILRB4 and IL-10Ra, CLEC7A and RELB.

Heatmap analysis displaying the overlapped targets which were dis-regulated under tuberculosis infections among the Gene Expression Omnibus public datasets GSE20050 (clinic), GSE57275 (in vivo), and GSE52819 (cultured cells based).
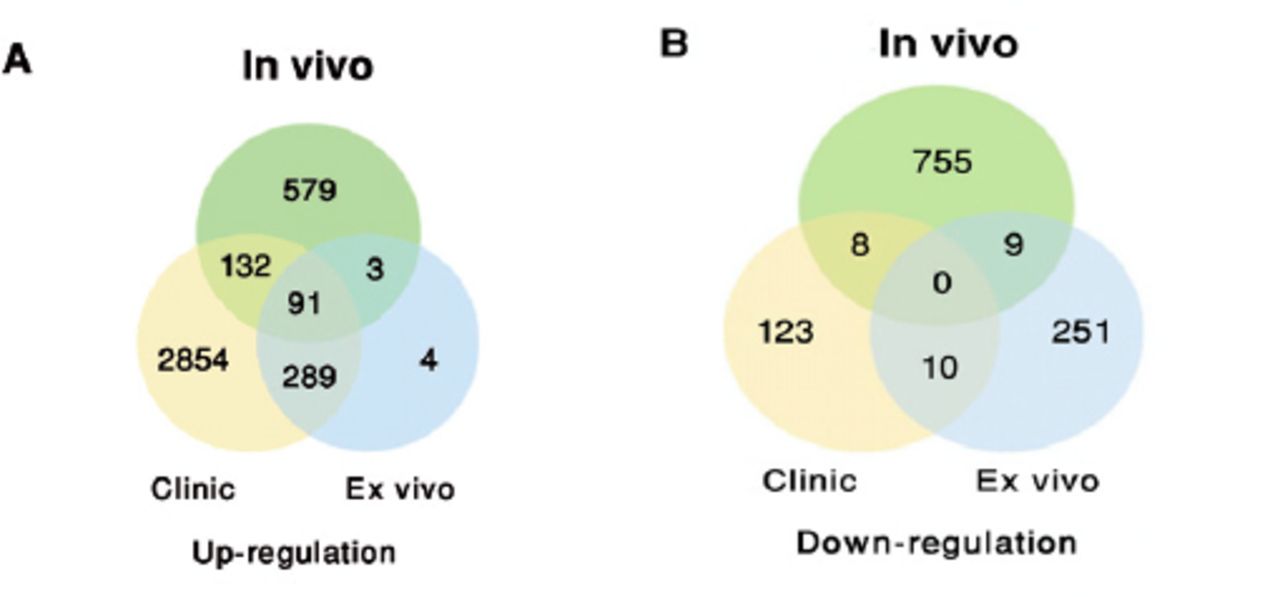
Venn diagram showing the distribution of the common A) up-regulated or B) down-regulated targets under Mycobacterium tuberculosis infections among the Gene Expression Omnibus public datasets.
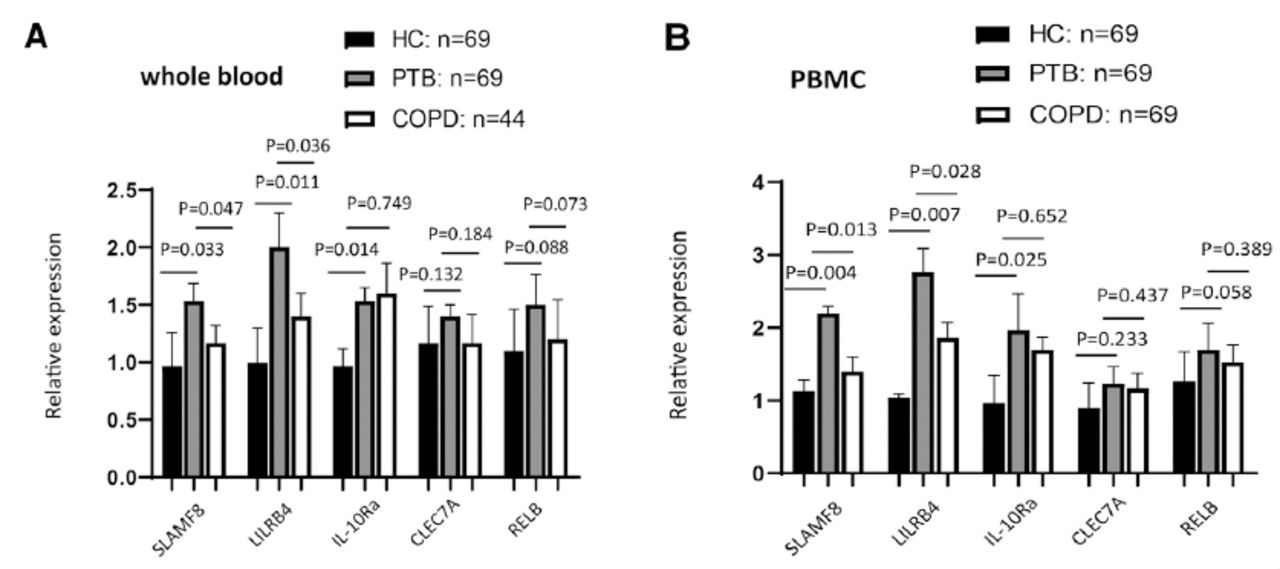
Preliminary validation experiments were conducted by qRT-PCR for 69 PTB patients, 44 COPD patients and 69 healthy controls using the samples of whole blood or PBMCs isolated ex vivo from the blood, which was not having an infection in vitro. The clinical information background for these subjects has been shown in Table 1. The results of our study demonstrated that the expression of SLAMF8, LILRB4, and IL-10Ra proteins was induced in whole blood (Figure 3) and PBMCs (Figure 4) of PTB patients as compared to the healthy control groups, and SLAMF8 and LILRB4 expressions could distinguish between PTB and COPD subjects. Meanwhile, there was no significant difference between the expression of CLEC7A and RELB in PTB patients and healthy control group.
Clinical characteristics of pulmonary tuberculosis patients and healthy volunteers.
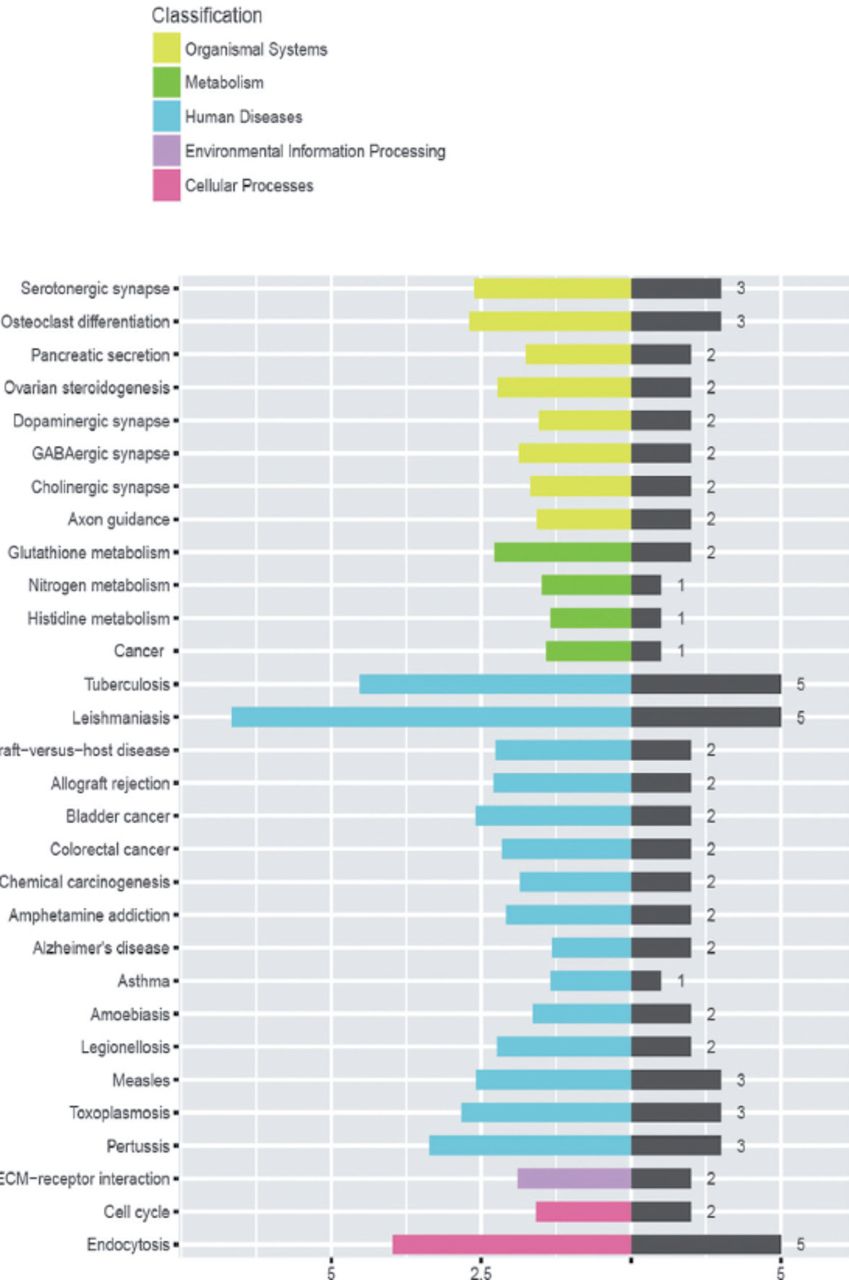
Kyoto Encyclopedia of Genes and Genomes bioinformatics analysis determined the pathways related to the differentially expressed genes identified in Figure 1.

Gene Ontology bioinformatics analysis enriched the biological functions related to the differentially expressed genes in Figure 1. FDR: false discovery rate
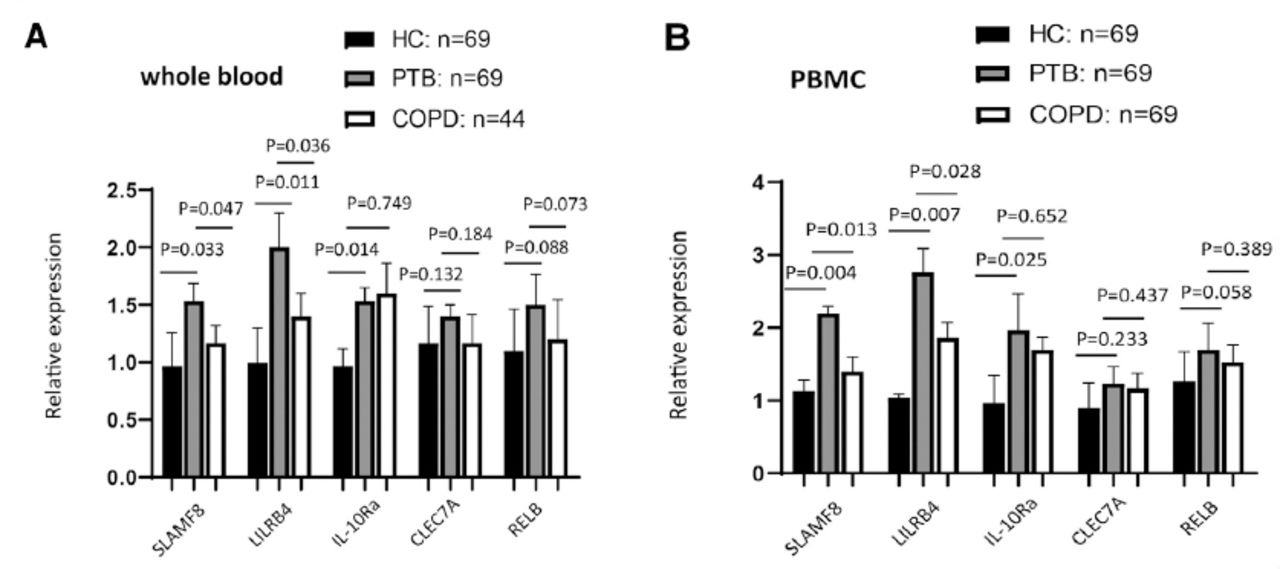
Validated the enhancement of the targets that were involved in negative regulation of immune system process in A) whole and B) peripheral blood monocular cell between active pulmonary tuberculosis (PTB) patients and healthy controls using reverse transcription polymerase chain reaction. COPD: chronic obstructive pulmonary disease
Verification of up-regulated protein abundance using PBMC from PTB patients
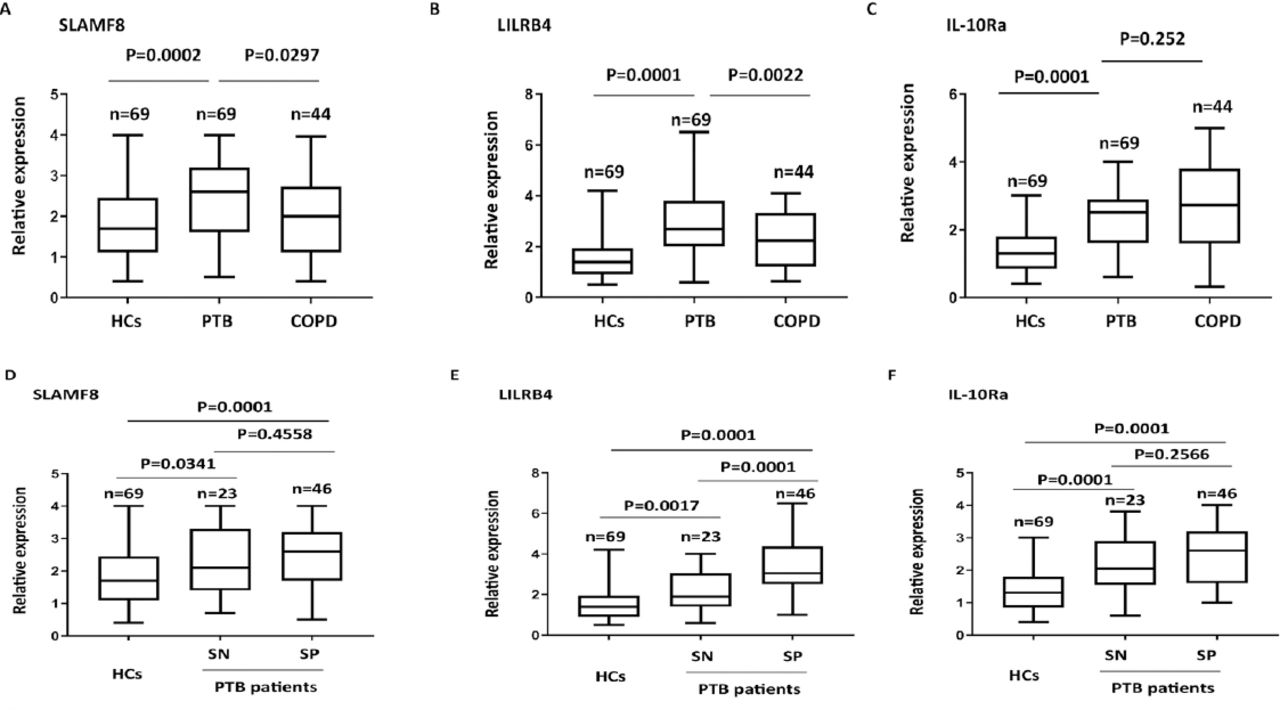
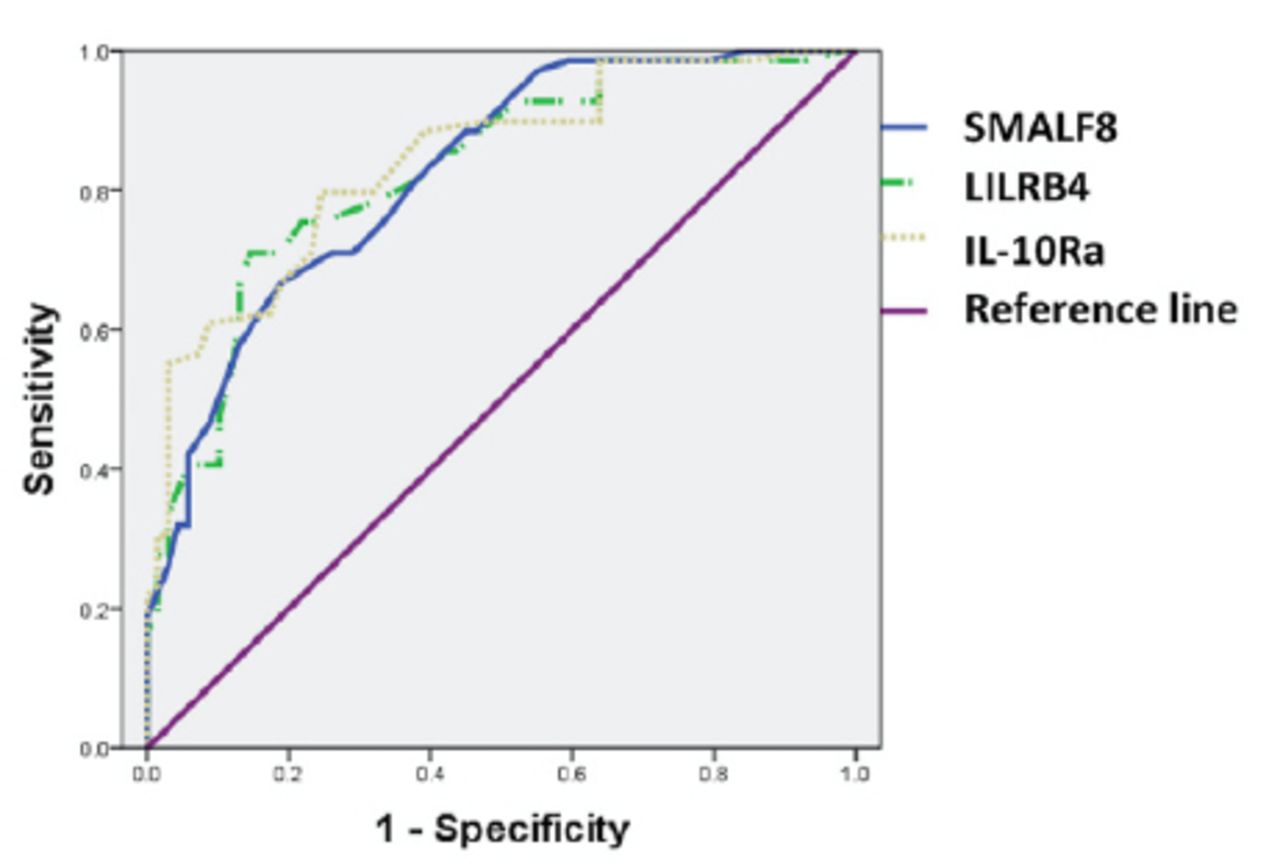
The changes in protein abundance for SLAMF8, LILRB4, and IL-10Ra in PBMC samples from PTB patients were further determined using ELISA. The abundance of SLAMF8, LILRB4 and IL-10Ra was significantly higher in PTB patients as compared to the control group; also, SLAMF8 and LILRB4 expressions could distinguish between PTB and COPD subjects (Figures 6A-C). Subgroup analysis showed that both smear positive and smear negative PTB patients had higher SLAMF8, LILRB4, and IL-10Ra expressions than the controls. In PTB patients, the level of LILRB4 was significantly higher in smear positive group as compared to the smear negative group; however, no significant difference was observed between the smear positive and smear negative groups for SLAMF8 and IL-10Ra (Figures 6D-F). Using the receiving operating curves (Figure 7), we obtained the areas under the curve values, optimal cut-off points, positive predictive value, and negative predictive values for the 3 targets and the corresponding sensitivity and specificity values for PTB diagnosis (Table 2).
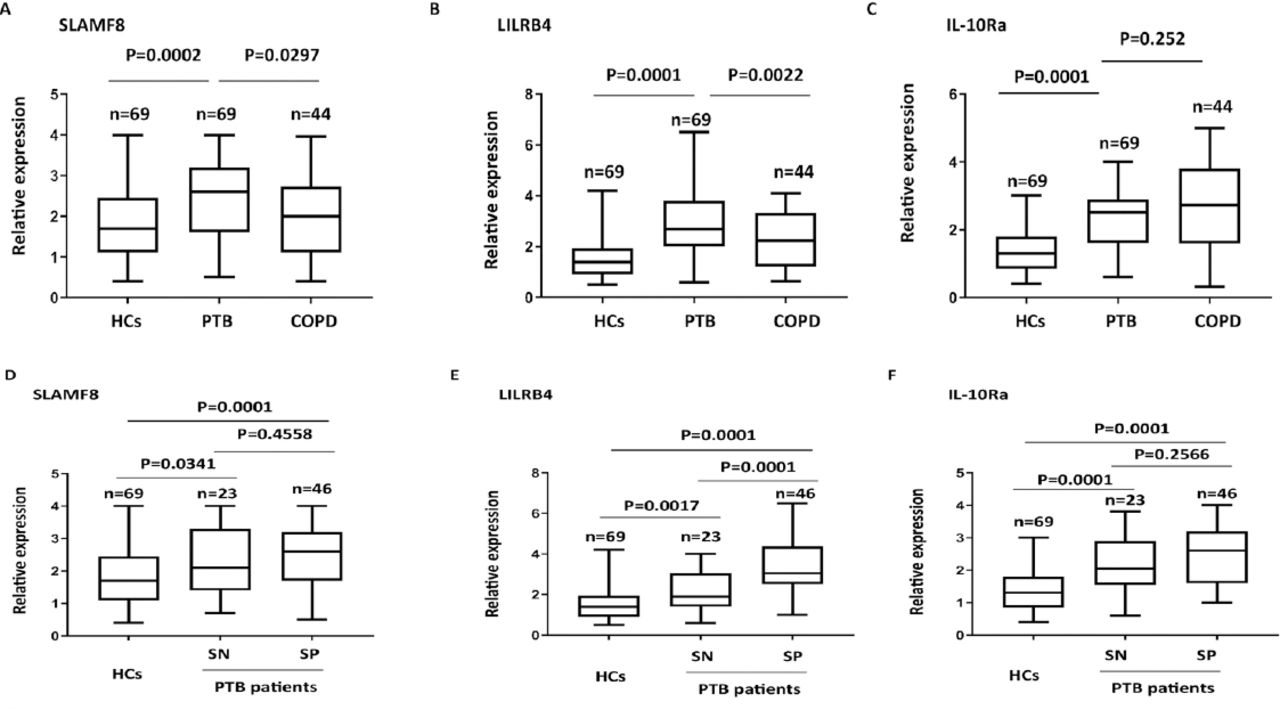
The protein abundance of A) SLAMF8, B) LILRB4, and C) IL-10Ra in different subgroup of pulmonary tuberculosis patients (PTB) with COPD and control subjects by using ELISA assay. Determination of the relative of D) SLAMF8, E) LILRB4, and F) IL-10Ra expressions in smear negative and smear positive PTB patients by using enzyme-linked immunosorbent assays assay. HC: healthy control, SP: smear positive, SN: smear negative

The receiving operating curve curve for the expressions of SLAMF8, LILRB4, and IL-10Ra in relation to the pulmonary tuberculosis patients (PTB).
Diagnostic value of SLAMF8, LILRB4, and IL-10Ra expressions to predict pulmonary tuberculosis.
Potential of SLAMF8, LILRB4 and IL-10Ra expression to predict the treatment outcome in PTB patients
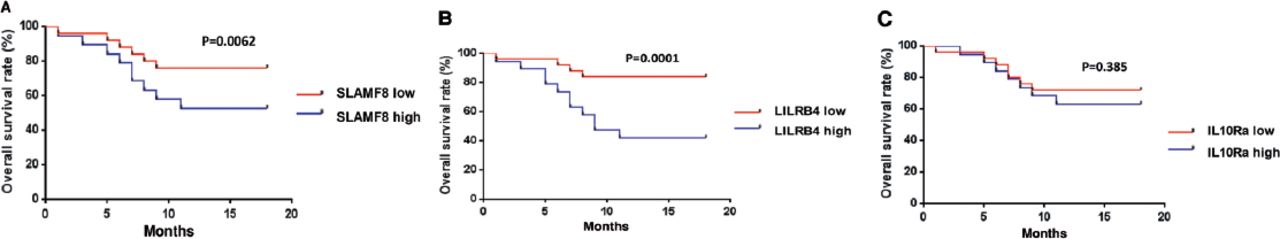
Further, we evaluated the potential of SLAMF8, LILRB4, and IL-10Ra protein abundance in predicting death rate within 180 days (6 months) of treatment. Patients were divided into 2 groups according to the median level of SLAMF8, LILRB4, and IL-10Ra. Subsequently, Kaplan-Meier analysis was carried out and it was found that SLAMF8 and LILRB4 groups with high expression showed significantly worse survival as compared to the groups with low expression (Figures 8A-B). In contrast to this, no significant difference was observed between the survival rates of high and low expression groups of IL-10Ra (Figure 8C).
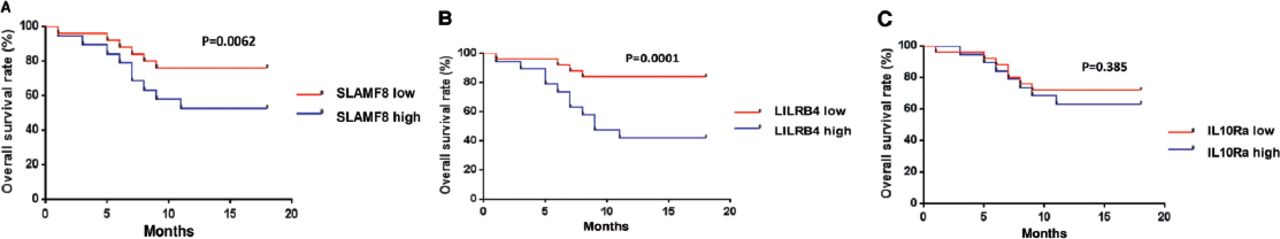
Kaplan-Meier curves of patients with tuberculosis produced according to the protein abundance of A) SLAMF8, B) LILRB4, and C) IL-10Ra expression. The p-values were determined by the log rank test.
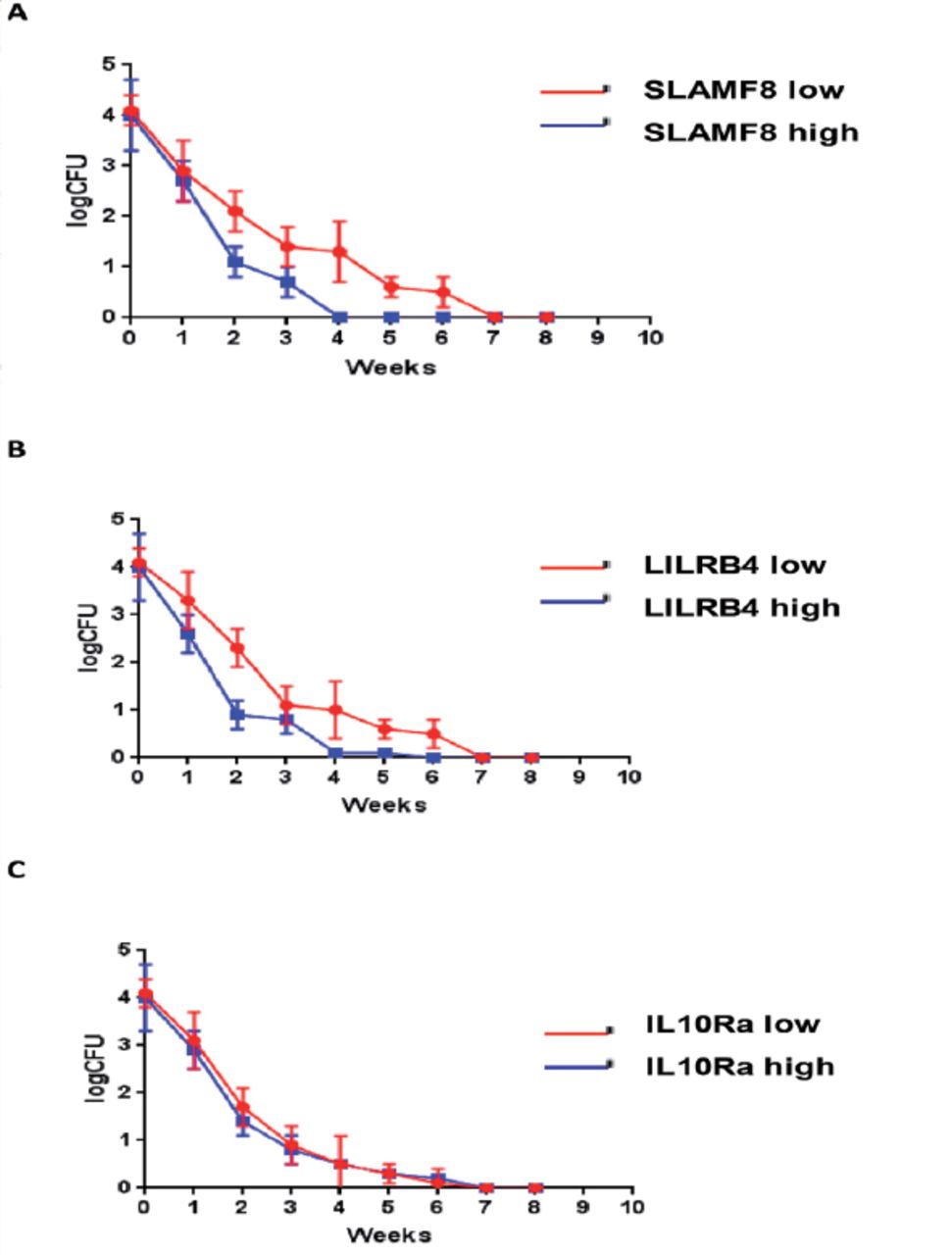
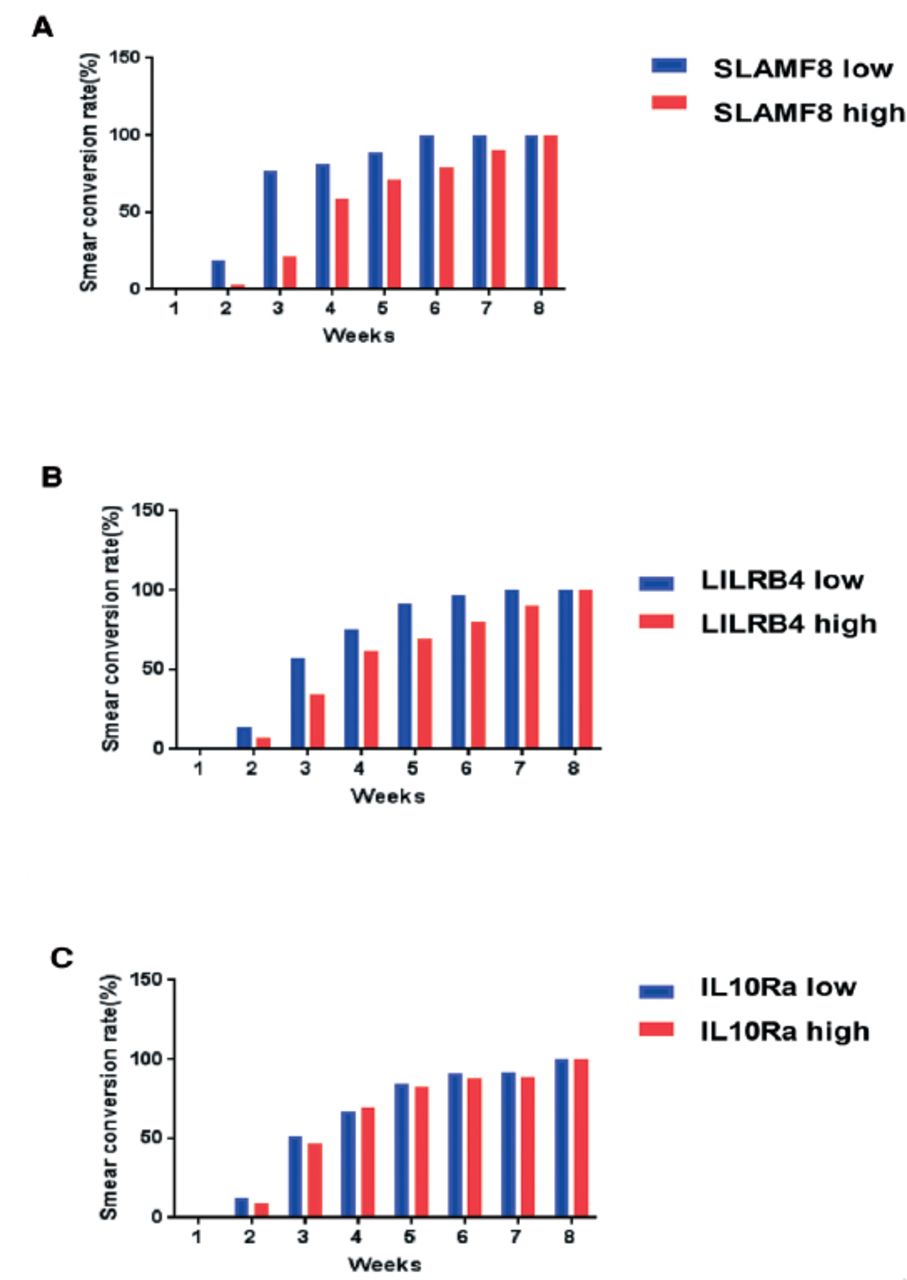
The M. tuberculosis was detected in sputum samples of 46 PTB patients out of 69 using the laboratory techniques (culture positive and smear positive). By monitoring the bacterial load of tuberculosis from these 46 patients, we found that the sputum CFU count of all patients decreased rapidly after 8 weeks of treatment indicating the good treatment outcome. While comparing the different groups of patients, it was found that the rate of decline was slower in the SLAMF8 and LILRB4 groups with high expression (Figure 9). Likewise, the rate of acid-fast conversion in the high expression groups of SLAMF8 and LILRB4 was significantly slower as compared to the low expression groups of SLAMF8 and LILRB4 at third week (Figure 10). To summarize, the results of our present study demonstrated that the identified targets for instance, SLAMF8 and LILRB4 may act as novel and promising biomarkers for the diagnosis and treatment outcome of PTB.

Weekly bacteriologic sterilization according to the protein abundance of A) SLAMF8, B) LILRB4, and C) IL-10Ra expression, were evaluated as the colony forming units per milliliter sputum, in the first 8 weeks treatment of pulmonary tuberculosis patients.
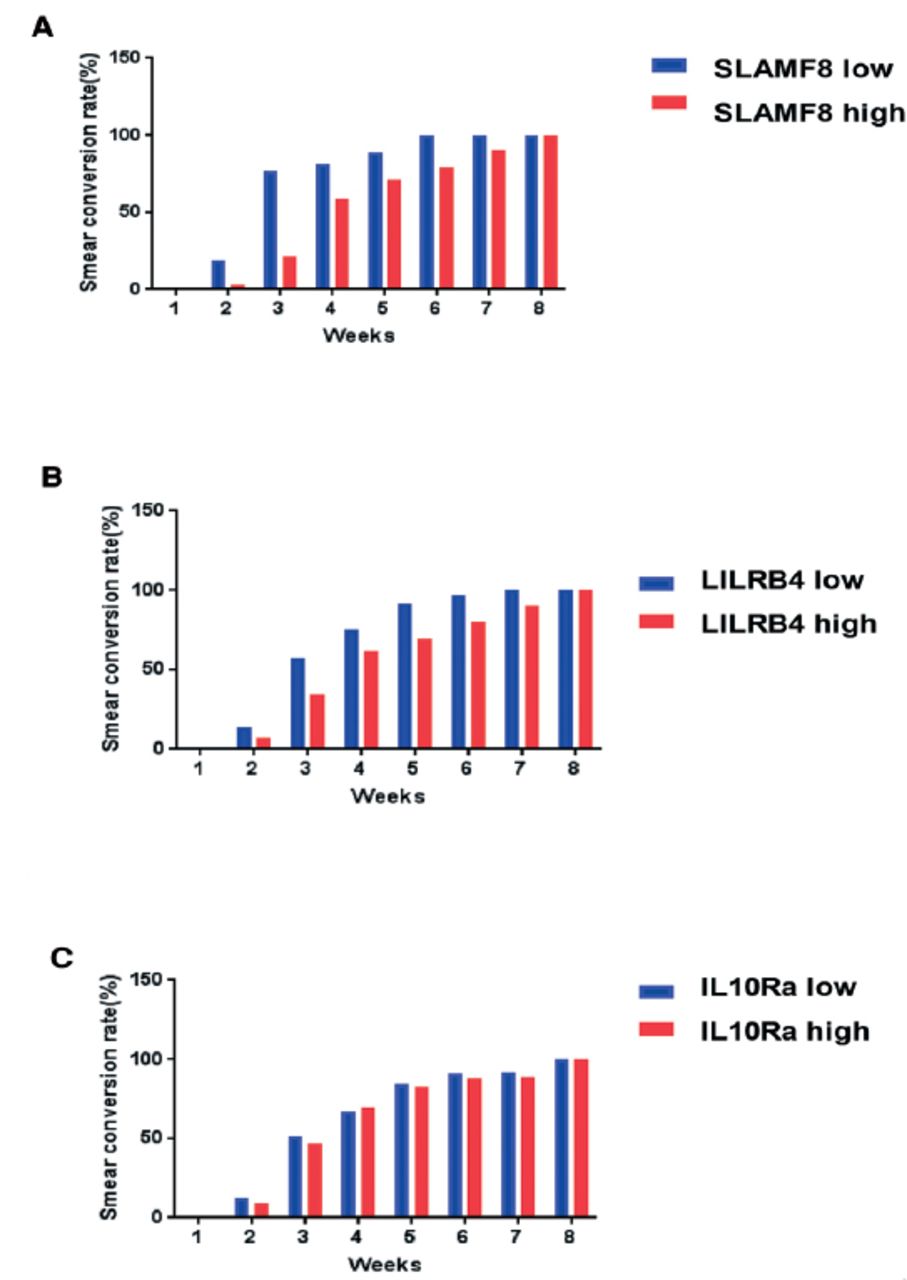
Weekly bacteriologic sterilization according to the A) SLAMF8, B) LILRB4, and C) IL-10Ra expression, were evaluated as the smear conversation rate, in the first 8 weeks treatment of pulmonary tuberculosis patients.
Discussion
Research has been carried out for development of biomarkers but only few have been taken successfully from the research laboratory into the routine clinical practice.4 The recent technological advances in genomics and proteomics have enabled researchers to discover novel biomarkers for various diseases. Walzl et al,4 reported the applications of mass spectrometry in identification of diagnostic markers of PTB.4 Preez et al,9 reported that metabonomics is a promising way to discover novel PTB biomarkers. Singhania et al,10 reported that transcriptomics is an effective tool for discovery of PTB biomarkers. Therefore, a large number of transcriptomics studies have been carried using the microarray and RNA-seq technologies for the analysis of TB infected samples.10 By analysis of the previously published datasets, we have found some novel targets such as SLAMF8, LILRB4 and IL-10Ra whose mRNA expression and protein abundance increased significantly in PTB patients, and decreased after PTB treatment. This suggests that these targets could dynamically reflect the severity of tuberculosis.
It is crucial to predict the treatment outcome and prognosis for individual PTB patients. Therefore, several studies have been carried out to develop and evaluate PTB’s predictive scores.11,12 Apart from this, ability of some biomarkers have also been investigated to effectively predict the treatment outcome of PTB. Wang et al,13 found vitamin D and the promoter metabolism to be closely related to the prognosis of PTB. Zheng et al,14 demonstrated that P2X7 gene polymorphism is a potential prognostic indicator of PTB. Some researchers have reported on the comprehensive use of high-through put screening technology and bioinformatics analysis to develop new prognostic markers of PTB.15,16 In the present study, 3 datasets of M. tuberculosis infection were used which represented different models and stages in the development of active PTB disease. Our results revealed that there was a significant change in the expression of proteins which were negatively related to immune response. The level of 3 identified proteins like SLAMF8, LILRB4, and IL-10R which were negatively related to immune response was found to be elevated in whole blood and PBMC samples active PTB patients as compared to the healthy controls. This is the first report util date which has identified these proteins as potential diagnostic markers of PTB. Interestingly, SLAMF8, LILRB4, and IL-10R act as cell surface receptors. SLAMF8 has been reported to restrict bacterial infection through regulating NADPH oxidase and reactive oxygen species pathway.17 Upon addition of SHP-1, LILRB4 down regulates macrophage activation signal mediated by non-receptor tyrosine kinase cascade.18 IL-10R complex consists of 2 different chains (IL-10Ra and IL-10Rb) and the combination of IL-10 and IL-10R mediates anti-inflammatory signal transduction.19 Previous reports have demonstrated that M. tuberculosis protects itself from human immunity during infection through several mechanisms.20 One mechanism involves the induction of proteins that were negatively related to immune response of infected macrophages which further helps in the maintenance of intracellular bacilli and inhibition of inflammation.21 Therefore, the highly expressed proteins that were negatively related to immune response could help with the immune escape of M. tuberculosis from the macrophage indicating poor treatment outcomes.22 Similar observations were made in our study which proved that the high expression of SLAMF8 and LILRB4 at the time of diagnosis resulted in poor treatment outcome as indicated by 6-month death rate and 8 weeks chemotherapy bacteriologic sterilization.
Study limitation
Although we found that the expression of SLAMF8 and LILRB4 may differentiate between PTB and COPD patients, we have not included other lung diseases as controls to determine the specificity of these newly discovered proteins for PTB. Further research is going on to explore the ability of SLAMF8, LILRB4, and IL-10Ra to distinguish PTB patients from that of non small cell lung cancer and brucellosis infection. Furthermore, the present study was conducted at only one hospital with small sample size. Therefore, further studies need to be conducted in longer time-frames using a large sample size at multi-centers.
In conclusion, the analysis of already known datasets indicates that the present study provides a novel promising way to identify novel biomarkers for the diagnosis and prognosis of pulmonary tuberculosis patients.
Acknowledgment
We would like to thank Editage (https://www.editage.com/) for English language editing.
Footnotes
Disclosure. This study was funded by the National Natural Science Foundation Project (Grant no. 81670009, 81702173).
- Received August 22, 2019.
- Accepted February 3, 2020.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.