


Abstract
Beta-thalassemia is a genetic disorder that is caused by variations in the beta-hemoglobin (HBB) gene. Saudi Arabia is among the countries most affected by beta-thalassemia, and this is particularly problematic in the Eastern regions. This review article is an attempt to compile all the reported mutations to facilitate further national-level studies to prepare a Saudi repository of HBB gene variations. In Saudi Arabians, IVSI-5 (G>C) and Cd 39 (C>T) are the most prevalent HBB gene variations out of 42 variations. The coinheritance of HBB gene variations with ATRX, HBA1, HBA2, HBA12, AHSP, and KLF1 gene variations were observed to be common in the Saudi population. National surveys on the molecular nature of hemoglobinopathies should be set up through collaborations between research centers from various regions to create a well-documented molecular data bank. This data bank can be used to develop a premarital screening program and lead to the best treatment and prevention strategies for beta-thalassemia.
Hemoglobinopathies are disorders that affect the hemoglobin molecule, which carries oxygen from the lungs to the rest of the body. Hemoglobin is a protein that consists of 2 α-like type and 2 β-like type chains. A variation on the globin genes can lead to an error in the production of the coded chains, which cause hemoglobinopathies, such as thalassemia and sickle cell diseases. Beta-thalassemia is a prominent recessive genetic disorder that is usually caused by single-point variations (formerly called mutations) or deletions that are inherited from parents who are carriers or affected by the disease variants. Sequence variations that completely prevent the synthesis of the β-globin chain of the hemoglobin molecule are termed β0 variants; however, variations that reduce protein expression or change the molecular nature of the protein chains are called β+ variants.1-3 The main objective of this review is to compile all previously reported β-hemoglobin gene (HBB) variants from the Saudi population to create a national HBB variation reference source for future research and clinical applications.
Saudi Arabia is among the Arab countries most affected by β-thalassemia. As a result, the Ministry of Health initiated a national program that provides premarital screening and genetic counseling to control the high prevalence of hemoglobinopathies by spreading awareness among nationals.4-6 The main reason for the high prevalence is the high percentage of consanguineous marriages among Saudis.7 The Ministry of Health requires that Saudis take a test if they want to get married. This screening test involves taking blood samples in ethylenediaminetetraacetic acid (EDTA) vacutainers from Saudis in designated centers around the country. The samples are then sent to one of the 125 specialized laboratories where hemoglobin electrophoresis and other standard blood testing is carried out.6-8 Those who have β-thalassemia or are carriers of the disease have a hemoglobin A2 (HbA2) level above 3.5% and a low mean corpuscular volume (MCV) with no appearance of iron-deficiency anemia.9,10 However, the use of the HbA2 level as a diagnostic marker for the identification of carriers or the β-thalassemia trait is debatable.11 Therefore, a precise country-wide data bank based on national-level research is needed to develop error-free molecular diagnostic methodologies.
Prevalence of b-globin mutations
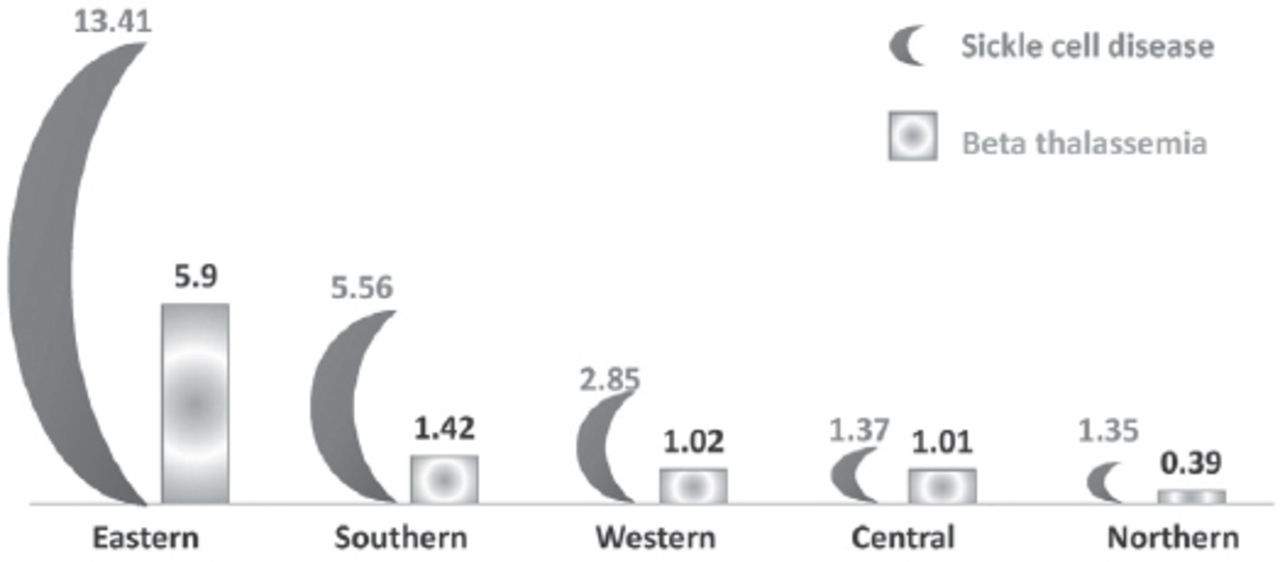
Studies on the molecular basis of thalassemia have been performed in a very limited number of Saudi Arabians. Except for the national study that was conducted in the 1980s and 1990s by El-Hazmi et al4,12,13 there has been no research on the molecular basis of β-thalassemia or on mutations that cause the disease in any region other than the Eastern Province and Makkah. In the last 20 years, only 6 studies were conducted on the molecular basis of β-thalassemia to determine the prevalence of β-globin gene mutations, whereas a few other studies reported only one or 2 HBB variations. Out of these 6 prevalence studies, 4 were conducted in the Eastern province and one was conducted every 5 years. The first study was conducted in 1999 by el-Harith et al14 and Borgio et al15 conducted the last study in 2016. Four different studies on the prevalence of β-thalassemia were conducted in 20 years, which is very low for a region with such high prevalence (Figures 1 & 2). Various studies have reported that the Eastern Province of Saudi Arabia had the highest prevalence of both β-thalassemia and sickle cell disease (Figures 1 & 2).2,16-20 However, according to the latest study on premarital screening and genetic counseling, which was performed in 2011, Jazzan, which is located in the southern region of Saudi Arabia, had the second highest prevalence of β-thalassemia in Saudi Arabia (Figures 1 & 2).8 However, no studies about the molecular basis of the disease in the Jazzan region have been conducted. The prevalence of sickle cell disease carriers is more than double the prevalence of β-thalassemia carriers in all regions of Saudi Arabia (Figure 1). No studies on the molecular nature of the hemoglobinopathies have been conducted on a national level in Saudi Arabia. Hence, this review article is an attempt to list all the reported mutations and combine them in a single paper. This data can be used to facilitate further national-level studies to create a national repository of all HBB gene variations.

Histogram shows the percentages of population who screened with HBB mutations either as homozygous or heterozygous (carrier/affected).

Map illustrates the spectrum of various HBB gene variations. It turns out that the Eastern Province and Makkah region which were with more number of mutations through prevalence studies in Saudis. Hufuf, Qatif, and Dammam are the centers where the reported mutations came from at the studies in the Eastern Province.
Etiology
The HbVar database contains more than 800 variations of the β-globin gene, including consensus splice sites variations, nonsense codon variations, splice junction variations, frameshift variations, cryptic splice site variations, variations at 5’UTR, nonsense codon variations and point variations (Table 1).21,22 Forty-two HBB gene variations were identified from various studies that were conducted either in Saudi Arabia or outside Saudi Arabia but with samples including Saudis.12,14,16,17,23-28 In this review, the mutations are listed in Table 2 in a specific order that shows how many studies reported the mutation. The Asian mutation IVSI-5 (G>C) tops the list, and the Mediterranean Cd39(C>T) mutation is second on the list (Table 2). The IVSI-5(G>C), Cd39(C>T), IVSII-1 (G>A), and IVSI-25bp mutations were reported in every prevalence study of β−thalassemia in Saudi Arabia (Table 2). The oldest record of a β-thalassemia mutation detected in a Saudi patient was from a study conducted abroad in 1986 by Boehm et al.23 This study characterized the Cd37 (G>A) mutation in a Saudi family. The IVS1-128 (TAG>GAG) mutation was the second oldest mutation to be characterized and was found in a Saudi patient in a study performed by Wong et al.24 El-Hazmi et al12 then initiated the first prevalence study of β-thalassemia-related gene variations in Saudi Arabia by identifying 14 β-thalassemia-causing mutations that were reported from around the country. In addition to these studies, a few other non-prevalence studies reported a few HBB gene variations among Saudis.25,28
A study conducted in the Jeddah region reported the highest number of variations (28 variations) in a single study.26 Also, a recent prevalence study performed by Borgio et al15 reported 6 mutations that were observed for the first time in Saudi Arabia in addition to 3 novel mutations (Tables 1 & 2). The intronic variations HBB:c.315 + 26T>G [IIVS+26(T>G)], HBB: c.315 + 81C>T [IVSII+81(C>T)], and HBB:c93- 23_94del (25-bp deletion) were found only in Saudi populations.2
Nature of HBB gene variations identified in Saudis.
List of mutations reported in Saudi Arabia or in Saudis anywhere else.
There were 24 β0 mutations (complete lack of β-globin protein production), 9 β+ mutations (reduced production of functional β-globin protein), 5 βi mutations (intronic mutations), and 2 β++ mutations (silent) found in the Saudi population (Table 1). All of the different types of HBB gene variations were reported from Saudi Arabia (Table 1). Frameshift HBB gene sequence variations were the most prevalent mutations among the β-thalassemia mutations in the Saudi Arabian population. The second most prevalent type was splice junction mutations, which hit the invariants GT or AG with 5 mutations. Only 2 mutations originated from Saudi Arabia [Cd37 (G>A) and IVS1-128 (T>G)], whereas most of the existing variations in Saudis were of Mediterranean origin (13 variations).29 Seven were from Asian India, 4 from the north neighbors Turkey and Kurdistan, and the rest originated from different parts of the world.
Co-inheritance
The co-inheritance of other gene (ATRX, HBA1, HBA12, and AHSP) mutations with HBB gene mutations was reported in Saudis (Figure 3).2,18,19,28 Recent studies have reported the co-inheritance of HBB gene variations with ATRX gene variations, such as c.485-58_485-55delAATG, c.662+65A>G, c.623delA, and c.848T>C. These 4 ATRX gene variations were reported to be novel and have been identified only in the Saudi Arabian population thus far.19 Out of these 4, 2 were exonic (on exon 8 and exon 9) in nature, and the authors reported a family history of developmental disability. It would be more appropriate to conduct a national-level survey on the co-inheritance of other gene mutations with HBB gene mutations. Because of the high rate of consanguinity in the Saudi population, the inclusion of screening for mutations in the ATRX gene, followed by proper genetic counseling, may decrease the prevalence of offspring with inherited genetic disorders.Studies on the co-inheritance of HBA1, HBA2, and HBA12 gene variations along with HBB gene variations have been performed in the Saudi population.18,28 Akthar et al18 reported the co-inheritance of 12 different α-globin gene variations in transfusion-dependent β-thalassemia major patients in the Eastern Province of the Kingdom of Saudi Arabia. In the healthy population, the -α3.7 deletion was reported to be the highest among the 12 different co-inherited α-globin gene variations in β-thalassemia major patients.18 The co-inheritance of different α-globin gene variations with the β-globin gene variations significantly reduced the levels of alanine transaminase and aspartate transaminase and the percentage incidence of osteoporosis, fracture, and splenectomy among the transfusion-dependent β-thalassemia major patients in the Eastern Province of the Kingdom of Saudi Arabia.18

Illustration for the co-inheritance of other gene mutations with HBB gene mutations. The outer grey circle imbedded with a tri-divided colored circle represented 3 HBB gene variations. Each one of the HBB mutation segment overlap exhibits the co-existence of other mutations that floated above them.
Very recently, there was a report on the co-inheritance of KLF1 gene variations [c.304T>C; c.544T>C; c.*296G>A; and c.*277C>G] with HBB gene variations (c.20A>T; c.25_26delAA; IVS I-5 (G→C); c.9T>C; c.17_18delCT; c.2T>C; NG_000007.3: g.71609_72227del619; c.46delT and c.93-23_94del) among β-thalassemia carriers from the Saudi Arabian population. The authors claimed that the 4 variations on the KLF1 gene (c.304T>C; c.544T>C; c.*296G>A; and c.*277C>G) were not significantly associated with borderline HbA2 among Saudi β-thalassemia carriers.30 The co-inheritance of variations at AHSP genes (AHSP:c.167T>G; AHSP:c.45G>T; AHSP:c.231G>T; and AHSP:c.168G>T) with HBB gene variations was also reported in the Saudi population.2
In conclusion and future research
β-hemoglobin gene variations, such as IVSI-5 (G>C) and Cd 39 (C>T), were the most prevalent among 42 variations that were reported in Saudis. The co-inheritance of HBB gene variations and variations in other genes, such as ATRX, AHSP, HBA1, HBA2, HBA12, and KLF1 were observed to be common in Saudi Arabians with β-thalassemia. Studies on the prevalence of β-thalassemia in Saudi Arabia were centered in the Eastern Province due to the high prevalence of hemoglobinopathies in that area. However, compared to the number of world-wide studies, the number of extant studies is still considered insufficient. No molecular studies have been performed in the region of Jazzan, which has the second highest prevalence of β-thalassemia. The region of Makkah does not have a high prevalence of hemoglobinopathies, but, because it is the site of many research institutions in Saudi Arabia, many studies on β-thalassemia have been conducted there. This review suggests that an affected region, such as Jazzan, should have a molecular research laboratory for hemoglobinopathies. The actual reasons for the presence of borderline HbA2 in Saudi β-thalassemia carriers remain unclear.31 To identify all the pathogenic variants, DNA sequencing-based molecular diagnosis protocols should be included in the existing premarital screening program. No recent studies on the molecular basis of hemoglobin-related disorders have been conducted on a national level in Saudi Arabia. Therefore, it is highly recommended that Kingdom-based national surveys on the molecular nature of blood disorders be arranged through collaborations between research centers from various regions to create a well-documented molecular data bank of blood disorders. Such studies can reveal the effect of co-inheritance on hemoglobinopathies and lead to the development of the best treatment and preventive strategies for β-thalassemia. Furthermore, a very recent study on the level of HbA2 and the presence of the HBB mutation proved that the level of HbA2 is not a diagnostic parameter for the identification of β-thalassemia carriers.31 Hence, the listed HBB variants can be identified in individuals through proper and simple technologies that minimize false positive and false negative results.
Acknowledgment
Authors would like to thank the Dean, Institute for Research and Medical Consultations (IRMC), Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia for her continuous support and encouragement. The author MAA wishes to express his appreciation to the Dean, IRMC, for giving a chance to work as volunteer at the Department of Genetic Research, IRMC. Authors thank Mr. Ranilo. M. Tumbaga, Mr. Horace T. Pacifico, and Mrs. Jee E. Aquino for their timely support. Line drawing of the map of figure 2 was drawn by Dr. Sayed AbdulAzeez and Dr. J. Francis Borgio.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.