


Abstract
Oral-facial-digital syndrome type I (OFDI) is an X-linked syndrome, which has several craniofacial and limb features; and hence, patients frequently present to craniofacial and plastic surgeons. Oral-facial-digital syndrome type I is caused by mutations in the CXORF5 gene. The gene product is one of the basal body proteins of a slim microtubule-based organelle called the “primary cilium”. Most of the clinical features of OFDI patients are related to dysfunctions of the primary cilium leading to abnormal Hedgehog signal transduction, depressed planar cell polarity pathway, and errors in cell cycle control.
Oral-facial-digital syndrome type I (OFDI) is inherited as an X-linked dominant trait, lethal in males. Its incidence is approximately one in 50,000 live births.1,2 Affected patients have craniofacial features (forme fruste median cleft lip, cleft palate, bifid/lobulated tongue, lingual hamartomas, teeth abnormalities, hypertelorism, brittle scalp hair, alopecia, milia of face/ears, hypoplasia of ala of the nose, central nervous system malformations, and mental retardation); limb anomalies (brachydactyly, clinodactyly, syndactyly, and rarely polydactyly of hands and feet); and cyst formation of internal organs (the brain, kidneys, liver, and pancreas).3-11 The syndrome is caused by mutations of the CXORF5 gene.12 Mutations in the same gene also cause Joubert syndrome and Simpson-Golabi-Behmel syndrome. We aim to review the pathogenesis of the clinical features of OFDI to enhance our understanding of the pathogenesis of craniofacial deformities, median clefts, and various limb features of OFDI patients.
Localization of the CXORF5 gene product
The OFDI protein is present in 3 main areas within the cell organelles: the centrioles of the mitotic spindle, the nucleus, and the “primary cilium”. Centrioles/centrosomes are involved in the mitotic spindle of cell division. Each centriole is a barrel-shaped structure made of 9 triplets of microtubules. The centrosome is made up of one pair of centrioles, which are surrounded by an amorphous mass of dense material (pericentriolar material). Singla et al13 showed that OFDI protein controls the length of centriole, is required for microtubule stability, and is important in recruiting intraflagellar transport protein 88 (IFT 88) to the centrosome. In the nucleus, the DNA combines with chromatin proteins forming the chromatin. This combination serves several functions, such as the package of DNA into a smaller volume and the prevention of DNA damage. During the interphase of the cell cycle, chromatin becomes loose to allow access to RNA/DNA polymerases for transcription/translation. Giorgio et al14 showed that OFDI protein localizes to the nucleus through physical interaction with subunits of the chromatin remodeling complex.
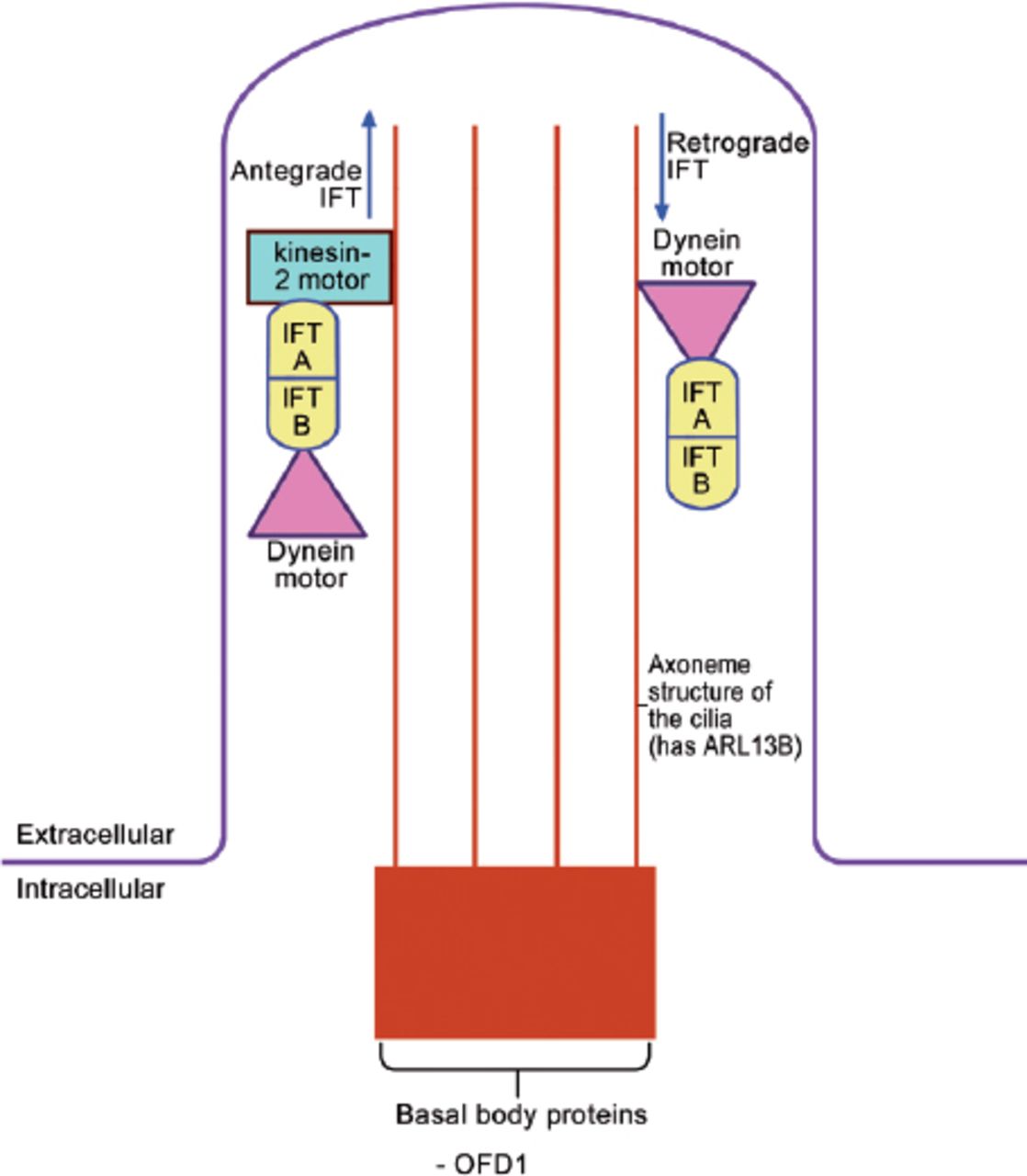
The “primary cilium” is a slim microtubule-based organelle that projects from the surface of many embryonic cells (Figure 1).15 It is composed of 3 parts: the basal body (a base that is attached to the apical actin network of the cell), the axoneme (a projecting part, which is made of 9 microtubule doublets with, or without a pair of central microtubules), and the ciliary membrane (the cell membrane around the axoneme). Oral-facial-digital syndrom type I is one of the basal body proteins (Figure 1). Note should be given that there are many other basal body proteins, such as the BBsomes (mutations of BBsomes cause Bradet-Biedle syndrome), FTM (Fantom, mutations cause Joubert syndrome), MKSI (mutations cause Meckel syndrome type I), and EVC (mutations cause Ellis Van Creveld syndrome). As expected, all these syndromes have overlapping clinical features with OFDI syndrome, and hence, they are all known as human “ciliopathies”.

The structure of the primary cilium and the intraflagellar transport within the cilium. IFT - intraflagellar protein transport, ARL 13B - ADP-Ribosylation factor like 13B, OFD 1 - oral-facial digital protein I
The primary cilium is the site of intraflagellar protein transport (IFT) (Figure 1).15,16 The ciliary basal body influences trafficking of proteins to the cilia. Antegrade IFT occurs when the protein is transported from the base to the tip, and is mediated by “Kinesin-2 motor” and 2 IFT proteins (IFT A and B). “Dynein motor” is also attached to the IFT proteins and this will be required for the retrograde IFT. Various proteins are modified and/or activated during this transport.
Functions of the primary cilium
Most of the clinical features of OFDI patients are related to dysfunctions of the primary cilium. The primary cilium is involved in 3 main functions: the Hedgehog signal transduction; the balance between 2 WNT pathways; the canonical (beta catenin) pathway, and the non-canonical planar cell polarity (PCP) pathway; the pathways of cell cycle control are as follows:
A) The Hedgehog signal transduction and OFDI
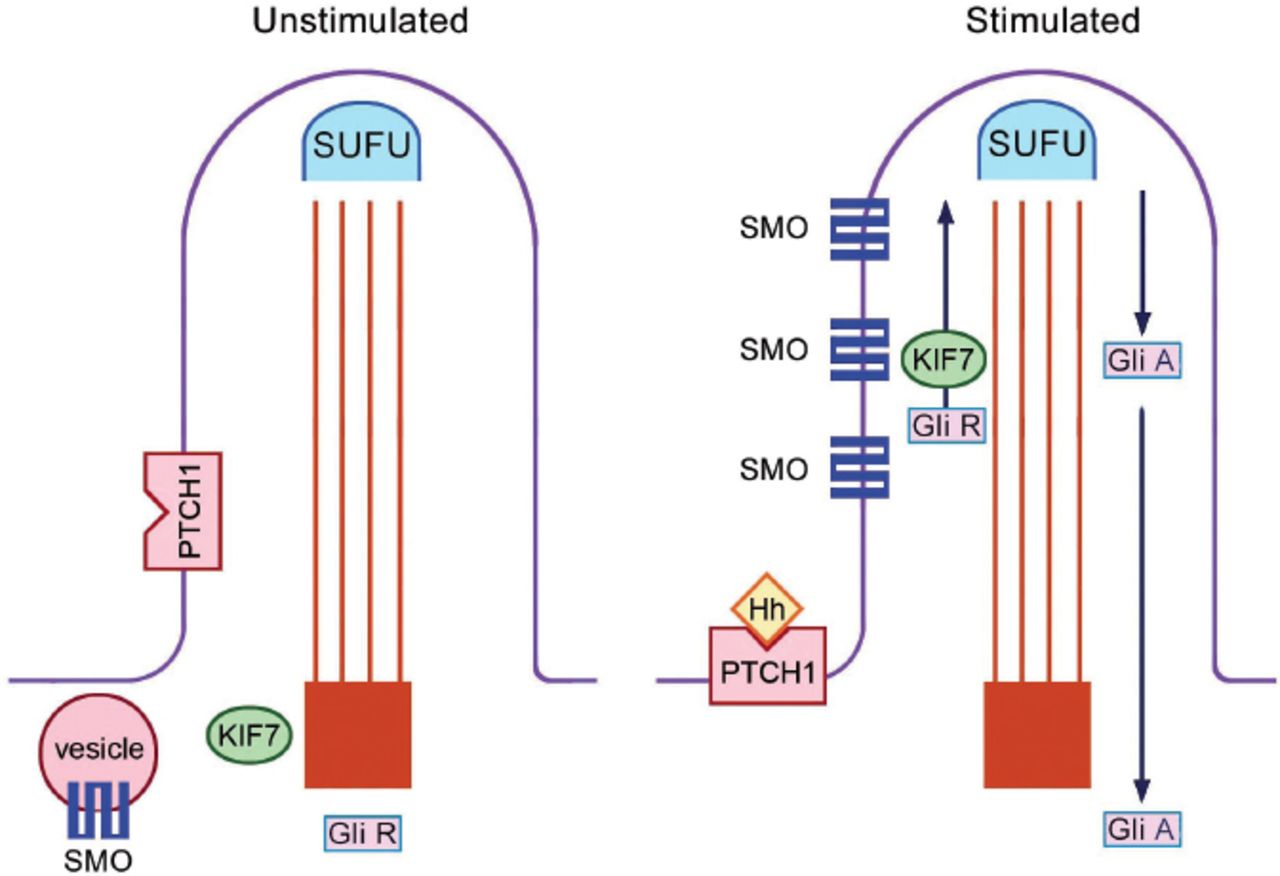
The primary cilium is the site of both Sonic Hedgehog (SHH) and Indian Hedgehog (IHH) signalling.15,16 The receptor for both SHH and IHH is called Patched 1 (PTCH1), which is located at the base of the ciliary membrane. In the absence of Hedgehog legends, the presence of PTCH1 at the base blocks smoothened (SMO). When the Hedgehog legend binds to PTCH1, the receptor moves away from the base of the cilium; and this allows entry of SMO to the ciliary membrane; allowing processing of proteins through the IFT (Figure 2). There are 2 forms of the GLI 3 protein: the activator (GLI 3A) and the repressor (GLI 3R) forms. GLI 3A is the full-length protein and GLI 3R is the short form. The GLI 3 activator/repressor ratio is dependent on SHH activation; the higher the SHH, the higher the GLI 3 activator/repressor ratio.

Hedgehog signal transduction within the primary cilium. In the unstimulated state, SMO is blocked. With Hh stimulation of the receptor PTCH1, SMO enter the cilium and intraflagellar protein transport is initiated. SUFU - suppressor of fused homolog (ensures efficient protein processing), PTCH1 - patched 1 (receptor for hedgehog), SMO - smoothened, KIF 7 - Kinesin family member 7 (involved in the up-going ante-grade protein transport), GLI - glioma-associated oncogene family zinc finger protein, GLI R - the repressor form of GLI, GLI A - the activator form of GLI, Hh - Hedgehog
(i) The neural tube and SHH in OFDI
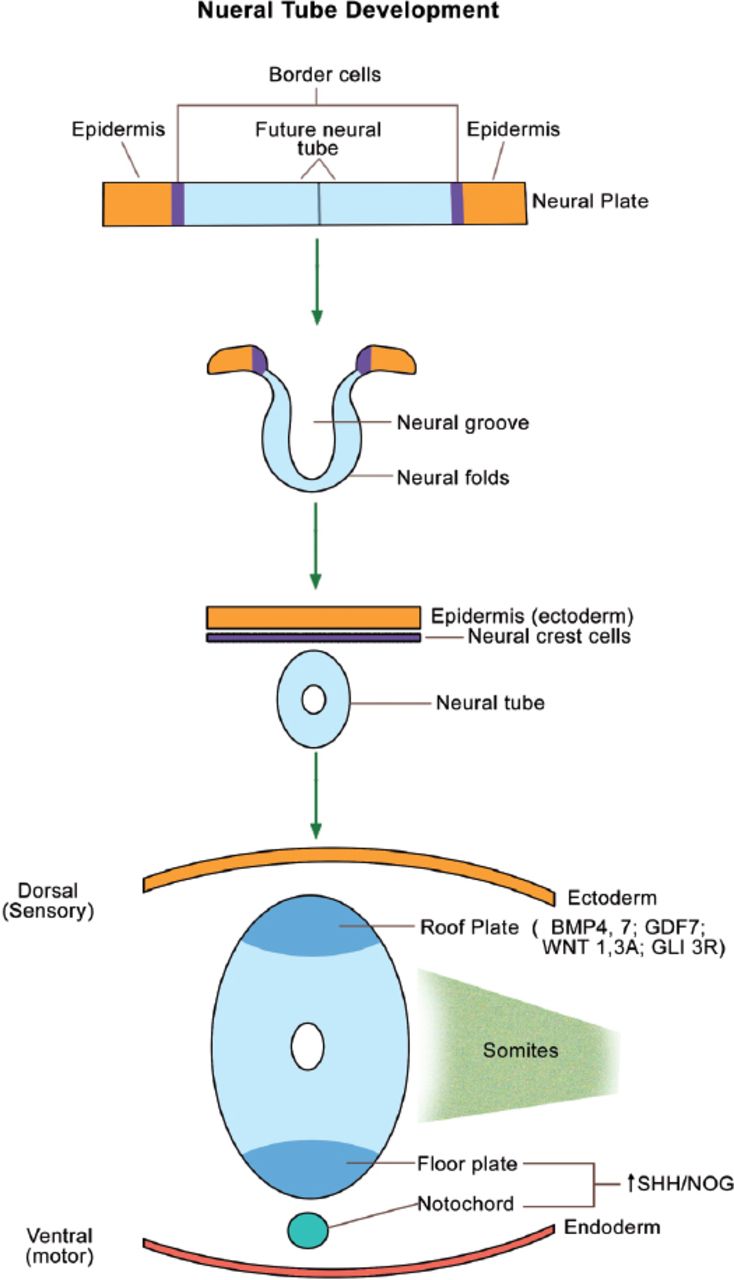
The neural tube forms the brain, spinal cord, and the nervous system. The neural crest cells (cranial ectoderm) will migrate to form the bones and cartilages of the face, as well as the teeth. The neural tube starts as a neural plate, which folds into a tube (Figure 3). The dorsal part of the neural tube (roof plate) forms the sensory neurons and expresses bone morphogenetic proteins 4 and 7 (BMP 4 & 7), growth and differentiation factor 7 (GDF 7), wingless mammary tumor virus 1 & 3A (WNT 1, 3A), and the repressor form of GLI 3 (GLI 3R). In contrast, the notochord (present ventral to the neural tube) and the ventral part of the neural tube (floor plate) form the motor neurons, and express SHH and NOG (NOG or Noggin is an inhibitor of BMPs). As expected, GLI 3 is processed in the primary cilia into the active form (GLI 3A) ventrally. The gradients (SHH/GLI 3A ventrally versus BMPs/GLI 3R dorsally) are essential for normal brain development/patterning (Figure 3).17 Cerebral/cerebellar abnormalities and mental retardation in OFDI patients18 are attributed to abnormal SHH signal transduction within the neural tube.

Neural tube development from the neural plate. Primary cilia are present wherever SHH is expressed (namely, the floor plate and notochord). BMP - bone morphogenetic protein, GDF - growth and differentiation factor, WNT - wingless tumor virus protein, GLI - glioma-associated oncogene family zinc finger protein, SHH - sonic hedgehog, NOG - noggin
(ii) The limb buds and SHH in OFDI
Sonic hedgehog is expressed in the posterior mesenchyme of the limb bud (in an area called the zone of polarizing activity).19 Similar to the neural tube, the SHH protein acts as a diffusible morphogen with a gradient. The highest concentration of SHH (and GLI 3A) is present in the posterior part of the upper and lower limb buds, and the concentration decreases as one approaches anteriorly. At the most anterior part of the limb bud, there is no SHH; and GLI 3R predominates. Abnormal SHH signaling (leading to a predominance of GLI 3R) leads to polydactyly,20,21 which is occasionally seen in OFDI patients. Sonic hedgehog has complex interactions with BMPs and fibroblast growth factors (FGFs).22,23 Syndactyly is frequently seen in OFDI patients. Syndactyly occurs secondary to errors of inter-digital apoptosis, which are partly mediated by BMPs and FGFs.19
Homeobox (HOX) genes are found in 4 clusters (HOX A, B, C, and D) in humans. There are 13 HOX genes in each cluster; but several of them are lost during development leaving a total of 39 instead of the expected 52. Individual genes are numbered according to their order in the cluster, with one being at the 3’ end and 13 being at the 5’ end of the cluster.24 Hence, the term “5’ HOX genes” means HOX 10-13. Sonic hedgehog induces a specific pattern of expression of the 5’ HOX genes, and this plays a role in digital patterning including digital length.24 Brachydactyly is the most frequently observed feature in the hands and feet of OFDI patients. Brachydactyly is known to be induced by errors of IHH, GDF5, BMP Receptor 1B, ROR 2 (tyrosine kinase receptor), and HOX D13.25 Ferrante et al,26 generated knock out animals lacking OFDI, and found altered expression of 5’ HOX A and 5’ HOX D genes in the limb buds. This suggests that the frequently observed brachydactyly in OFDI patients is probably related to errors of 5’ HOX gene expression.
(iii) The limb buds and IHH in OFDI syndrome
Indian hedgehog signal transduction also occurs in the primary cilium. Indian hedgehog is essential in coordinating chondrocyte proliferation/differentiation, and is mainly expressed in the pre-hypertrophic chondrocytes. Parathormone-related protein (PTHrP) helps mediates the effects of IHH through the formation of a biofeed-back loop.27 Interactions of FGFs, BMPs, IHH, and PTHrP integrate chondrocyte proliferation and hypertrophic differentiation.28 Abnormal IHH signaling in animals with ciliary dysfunction resulted in growth plate dysfunction and cartilage abnormalities.29 This suggests that the frequently observed clinodactyly and cone-shaped epiphysis10 in OFDI patients is secondary to errors of IHH signaling.
B) Oral-facial-digital syndrom type I and the balance between WNT pathways
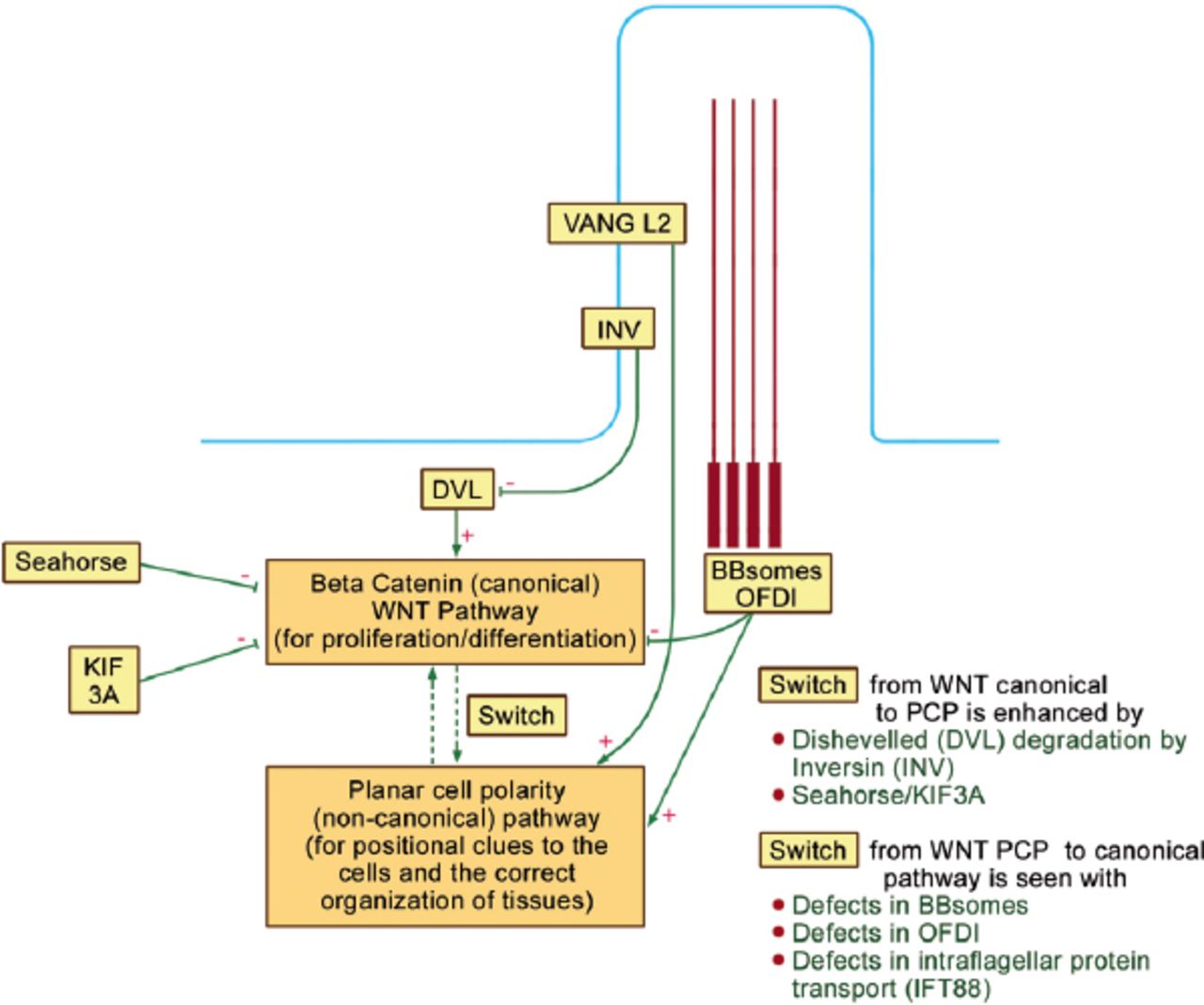
Wingless mammary tumor virus pathways are important for development, proliferation, and cellular orientation.22 Oral-facial-digital syndrom type I (and other basal body proteins, such as the BBsomes) are involved in the normal balance between 2 WNT pathways within the cell: the canonical (beta catenin) and the non-canonical PCP pathways (Figure 4).30 The former (canonical) pathway controls cell proliferation and differentiation, while the latter PCP is essential for positional clues to the cells including the proper orientation of the mitotic spindle and the correct organization of tissues. As seen in Figure 4, Dishevelled (DVL) is important for the canonical pathway, while VANGL2 (Vang Like 2 within the ciliary membrane) is important for the PCP pathway. Oral-facial-digital syndrome type I and other BBsomes enhance the PCP pathway, and depress the canonical pathway. Similarly, “Seahorse” and KIF3A (KIF3A is a subunit of kinesin 2 motor) depress the canonical pathway, and hence, enhance “switching” to the PCP pathway. Inversin (INV) is also expressed on the ciliary membrane and it degrades DVL, as well as, enhances “switching” to the PCP pathway.30

Oral-facial digital protein I (OFDI) is involved in the balance between wingless tumor virus (WNT) canonical and planar cell polarity (PCP) pathways. Dishevelled (DVL) is important for the canonical pathway, while Vang Like 2 within the ciliary membrane (VANGL2) is important for the PCP pathway. OFDI and other BBsomes enhance the PCP pathway and depress the canonical pathway. Similarly, “Seahorse” and a subunit of kinesin 2 motor (KIF3A) depress the canonical pathway and hence enhance “switching” to the PCP pathway. Inversin (INV) is also expressed on the ciliary membrane and it degrades DVL and hence it also enhances “switching” to the PCP pathway.
A suppression of the PCP pathway explains the median cleft of the upper lip, the median cleft palate, the alar hypoplasia, the bifid/lobulated tongue, and the hypertelorism in OFDI patients, because the PCP pathway is responsible for proper tissue migration and intercalation of cells in the embryonic midline.15,16
The upper lip develops at the inferior aspect of the fronto-nasal process. The neural crest condenses forming the nasal pit with the development of 2 medial and 2 lateral nasal processes. The maxillary processes of the first branchial arch migrate toward the medial nasal processes (Figure 5). The secondary palate and the lateral parts of the upper lip develop from the maxillary processes. The philtrum and pre-maxilla develop from the merging of the 2 medial nasal processes with each other toward the midline. The cleft lip in all reported cases of OFDI have been in the midline and are secondary to an error of the merging process in the midline. The lateral nasal processes also migrate toward the midline forming the nasal ala. The hypoplastic nasal ala seen in OFDI patients is also secondary to poor migration of the lateral nasal processes.

Merging toward the “midline” during the development of the cranio-facial skeleton.
The anterior two-thirds of the tongue develop from 2 lateral lingual swellings and the median tuberculum impar (from the first branchial arch). The 2 lateral swellings migrate and unite in the midline, and improper migration explains the bifid/lobulated tongue in OFDI patients. Similarly, incomplete migration of the orbits toward the midline may also explain the hypertelorism in these patients.
The exact pathogenesis of polycystic kidneys31,32 and fibrocystic disease of other internal organs, such as the liver and pancreas33 is unknown. One theory of pathogenesis is related to defective PCP.34-36 During normal kidney development, dividing cells in the renal tubules orient their mitotic spindles parallel to the lumen and thus, the net result of cell division is tubular “elongation” and not “dilation”. With PCP defects, mitotic spindles are miss-oriented leading to the formation of cysts.34-36
C) Defective cell cycle control in OFDI patients
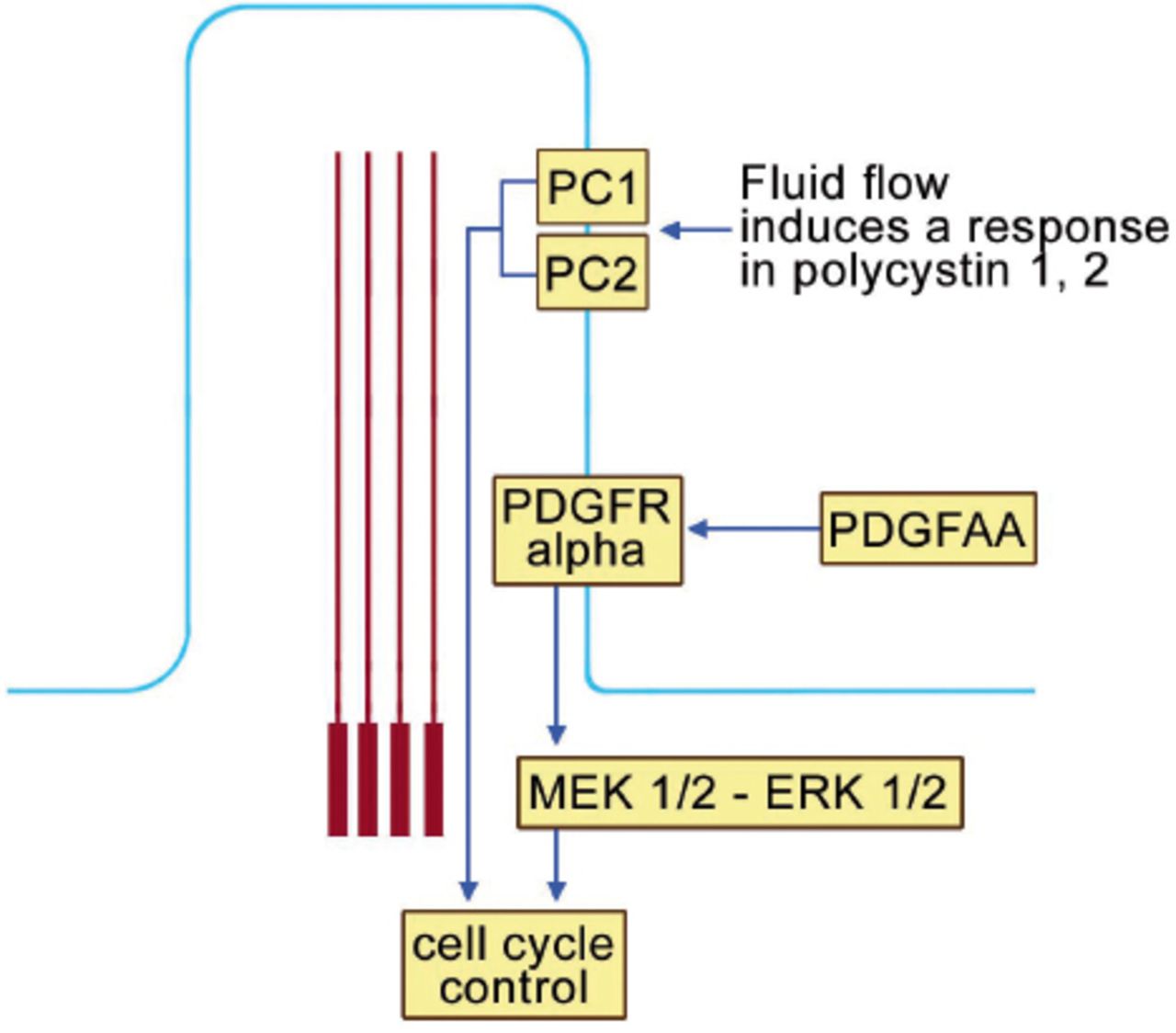
The primary cilium plays an important role in cell cycle control (Figure 6). Polycystin 1 and 2 (PC1 and PC2) localize to the ciliary membrane. In response to fluid flow, PCI and 2 are able to influence the cell cycle. Furthermore, the platelet derived growth factor receptor alpha (PDGFR Alpha) also localizes on the ciliary membrane. When platelet derived growth factor AA (PDGF-AA) binds into its receptor, secondary activation of (MEK ½ - ERK ½ (mitogen activated protein kinase ½ - extracellular signal regulated kinase ½) which, in turn, affects cell cycle control (Figure 6). Ciliary dysfunction of OFDI results in errors of cell cycle control and this participates in the pathogenesis of several of the clinical features of OFDI.

The primary cilium and cell cycle control. Polycystin 1 and 2 (PC1 and PC2) localize to the ciliary membrane. In response to fluid flow, PCI and 2 are able to influence the cell cycle. Furthermore, the platelet derived growth factor receptor alpha (PDGFR Alpha) also localizes on the ciliary membrane. When platelet derived growth factor AA (PDGF-AA) binds into its receptor, secondary activation of (MEK ½ - ERK ½ (mitogen activated protein kinase ½ - extracellular signal regulated kinase ½), which, in turn, affects cell cycle control.
The polycystic kidney disease genes (PKDI&2) encode PCI&2. Hence, abnormal cell cycle control is an important factor in the pathogenesis of cysts of internal organs.30 Similarly, abnormal teeth including the absence of the mandibular lateral incisors may also be related to abnormal cell cycle control.1,2 Primary cilia are present in hair follicles throughout morphogenesis and during hair follicle cycling after birth. Disruption of cilia of the hair follicles results in severe hypotrichosis because the follicles arrest at the second stage of hair development.37 Therefore, it is of no surprise that alopecia is a feature of OFDI. Milia are the result of cystic alterations of the trichoepitheliomatous bulge proliferation, and hence milia are also related to abnormal cell cycle control.38 Finally, lingual hamartomas are abnormal proliferations related to defective cell cycle control.
Discussion
The role of the vertebrate primary cilium in development, homeostasis, and disease is complex.39 In this paper, the literature on the pathogenesis of the clinical features of OFDI syndrome is reviewed and a summary is shown in Table 1. Abnormal Hedgehog signaling is responsible for neural and limb features. It is important to realize that SHH and IHH are both expressed in OFDI patients. However, trafficking of proteins and transcription factor activation (such as, GLI 1& 3) are deficient because of the mutated CXORF5 gene. This will also affect the complex interactions of SHH in the neural tube and limb bud, as well as the IHH interactions during cartilage and bone development. Animal models of OFDI reproduced the main features of OFDI syndrome in humans.26 However, polydactyly is almost a constant feature in mutant mice;26,40 whereas brachydactyly and clindodactyly are far more common than polydactyly in the human phenotype. This may be attributed to different interactions in animals and humans. Recent experiments showed that OFDI protein is not only involved in patterning of the brain, but also is involved in the regulation of neuronal differentiation of embryonic stem cells.41 This recent work will open new insights on the pathogenesis of brain anomalies and mental retardation in OFDI patients.
Summary of the pathogenesis of the clinical features of oral-facial digital protein I.
The PCP pathway is not only responsible for proper tissue/muscle spindle orientation, but also for intercalation of cells in the embryonic midline. CXORF5 mutations result in defects in this “midline” intercalation, and this is responsible for most of facial features, as well as, the bifid tongue deformity. It is interesting to note that migration to areas away from the “midline” is not affected. For example, the lateral lip component of the maxillary process migrates to meet the lateral aspect of the medial nasal process away from the midline. An error of such a migration will result in unilateral or bilateral cleft lip. The clefts of all reported cases of OFDI have been in the midline. In mice models, “split sternum” in the midline has also been observed.42 Finally, defective cell cycle control explains the formation of cysts of internal organs, milia (milia are cystic changes of the trichoepitheliomatous bulge), as well as some of the oral features, such as the missing teeth and lingual hamartomas (Table 1).
In conclusion, our review defines the pathogenesis of the clinical features of the OFDI syndrome to be related to dysfunctions of the primary cilium leading to abnormal Hedgehog signal transduction, depressed PCP pathway, and errors in cell cycle control.
References
* References should be primary source and numbered in the order in which they appear in the text. At the end of the article the full list of references should follow the Vancouver style.
* Unpublished data and personal communications should be cited only in the text, not as a formal reference.
* The author is responsible for the accuracy and completeness of references and for their correct textual citation.
* When a citation is referred to in the text by name, the accompanying reference must be from the original source.
* Upon acceptance of a paper all authors must be able to provide the full paper for each reference cited upon request at any time up to publication.
* Only 1-2 up to date references should be used for each particular point in the text.
Sample references are available from:
Acknowledgment
The authors would like to thank Mr. Virgilio Salvador, Medical Illustrator at King Khalid University Hospital, Riyadh, Kingdom of Saudi Arabia for drawing Figures 1-6.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company. This work was funded by the College of Medicine Research Center, Deanship of Scientific Research, King Saud University, Riyadh, Kingdom of Saudi Arabia.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.