


Abstract
Objectives: To assess completeness of reports in the Saudi Adverse Event Reporting System (SAERS), which is a part of the Saudi Food and Drug Authority pharmacovigilance system for monitoring the safety of medications.
Methods: A cross-sectional study was conducted in Riyadh, Saudi Arabia using the reports that were received between December 2009 and June 2012 in the SAERS. The completeness was assessed by reviewing the components of the adverse drug reactions (ADRs) form, and how many fields were completed. Descriptive statistics are reported.
Result: There were 14,783 reports during the study period. Eighty percent of these reports were spontaneous reports. Information related to the drug (99%) and adverse events (98%) of the reports were completed. While the patient’s demographic data were completed only in 38% of all reports, the least completed item in the ADRs form was the reporter information (15%). The most reported drug class was tumor necrosis factor inhibitors (7%), whereas events involving the respiratory organ system were the most frequently reported (4.5%).
Conclusion: Although the SAERS is considered new, it has a high number of reports. More efforts are needed to improve the completeness of the SAERS to be a good source to assess the signals between events and suspected drugs, especially when there is a high number of reports.
The Saudi Food and Drug Authority (SFDA) was established in March 2003. The Authority’s objective is to ensure the safety of food and drugs for humans and animals, and the safety of biological and chemical substances, as well as electronic products.1 In March 2009, the SFDA launched the Saudi Adverse Event Reporting System (SAERS) as a new pharmacovigilance system for monitoring the safety of the post-marketed medications.2 Pharmacovigilance is defined by the World Health Organization as “the science and activities relating to the detection, assessment, understanding, and prevention of adverse effects or any other drug-related problem”.3 Pharmacovigilance plays a major role in pharmacotherapeutic decision-making.4 Pharmacovigilance activities include actions to detect and assess adverse drug reactions (ADRs), evaluation of the probability of the association between the drugs and the ADRs, and actions taken in order to assure the safe use of medications.5 The SFDA pharmacovigilance system receives reports, submitted by health care professionals, manufacturers, or from consumers (patients) from all regions in Saudi Arabia, as well as internationally (especially for reports submitted by manufacturers).6 As with all regulatory bodies’ requirements for ADR submissions, the Saudi Pharmacovigilance System (SPS) has 4 mandatory fields to accept the Individual Case Safety Report (ICSR). These fields are: identifiable patient, suspect drug(s), event(s), and an identifiable reporter. Other important information is optional, such as patient age, comorbidity, concomitant use of medications, the date of the event, the date of recovery, and information on whether the event subsided while de-challenging (when the suspected drug was stopped) or if it reappeared after re-challenging (when the suspected drug was reordered). Complete and accurate information sent to the SPS will help the evaluators to investigate if there is a signal of association between a drug and ADRs. Therefore, the aim of this study was to assess the completeness of ADRs reports in the SAERS.
Methods
The study was conducted on the reports entered into the SAERS’s electronic database at the SFDA. The SAERS receives the ADRs reports from all regions in Saudi Arabia, as well as from international manufacturers. A confidentiality agreement between the investigators and the SFDA was agreed. The investigators had no access to the patients’ and reporters’ names or contact and company information, while reviewing and analyzing the reports in the SAERS database. This study was approved by the research committee at the Medication Safety Research Chair at King Saud University, Riyadh.
Study design
This was a cross-sectional study that included all reports to the SPS between December 2009 and June 2012. The pharmacovigilance center at the SFDA accepts all reporting forms (forms made by the SFDA or by hospitals) including the ‘ADRs for Health Professional form’, the ‘ADRs for Consumer form’, and the Council for International Organizations of Medical Sciences’ (CIOMS) form’. The latter form is used by pharmaceutical companies. These reporting forms have the same fields; however, the ADRs for the consumer form are available in the Arabic language.
The SAERS electronic data from the SAERS database were exported to the IBM SPSS Statistics for Windows version 20.0 (IBM Corp, Armonk, NY, USA), and then combined according to the SFDA identification number for the statistical analysis and evaluation of the different variables. Each report contains sections describing information related to the patient (age, weight, height, gender, institution), information related to the drug(s) (generic name, dose, route, start and end date, if the drug was suspected to cause the event or whether it was concomitant), information related to the event, the action taken after the event, the outcome of the event, the seriousness of the event, and information related to the reporter.
The reports were first assessed to verify they were valid for the study. To do this, all reports were reviewed to check if they had the minimum 4 mandatory fields that the SPS needs to accept the report: identifiable patient, suspect drug(s), event(s), and identifiable reporter. Next, the reports were assessed for completeness by reviewing all the elements in the ADRs reporting forms, and how many of these fields were completed.
For completeness, each field of the report elements was checked to find out whether it was completed, and the percentage of the completed fields was computed. Furthermore, these fields were stratified by different elements such as source of submission, type of profession, and other elements. For quality and adequacy, 2 independent reviewers reviewed 30 randomly selected reports to assess the quality of the information describing the adverse event. The adequacy of these reports was based on the criteria of a good quality report.7 The definition of a good quality report is that it should mention at least one of the 3 criteria: 1) if a specific event or adverse event was mentioned, 2) if the report mentioned signs or symptoms of a specific adverse event and included laboratory value or diagnostic tests and other information such as concomitant diseases or concomitant medications, and 3) if the reported signs or symptoms, and full details on the event and the patient.
Statistical analyses
Descriptive statistics for all elements of the SAERS were calculated. The age group of the reports were determined by computing the ‘age at onset of action’ with ‘the date of birth’. To determine the most reported drugs and event type each year, the statistics were carried out according to the ‘event onset of action date’ of each report. The data was analyzed using the IBM SPSS Statistics for Windows version 20.0 (IBM Corp, Armonk, NY, USA) except data related to the frequency of the event, age group, and type of institution were analyzed by using the Statistical Analysis Software (SAS), version 9.2 (SAS Institute Inc., Cary, NC, USA)
Results
The total number of reports received by the pharmacovigilance center and available at SAERS between December 2009 and June 2012, were 14,783. During the study period, the number of reports increased dramatically. Since the reports did not contain ‘report date’, the analyses were based on the event date. The total reports containing event date were 7,226 reports; of these, 439 reports (6%) were received in 2009, while 816 (11%) reports were received in 2010, and 1826 (25%) in 2011. In 2012, the number of reports increased to 4,145.
In the study period, the reports included a total of 44,787 events involving a total of 48,417 drugs. Each report involved a mean of approximately 3 events (range: 1-61) and 3 drugs (range: 1-22).
Assessing the completeness of SAERS
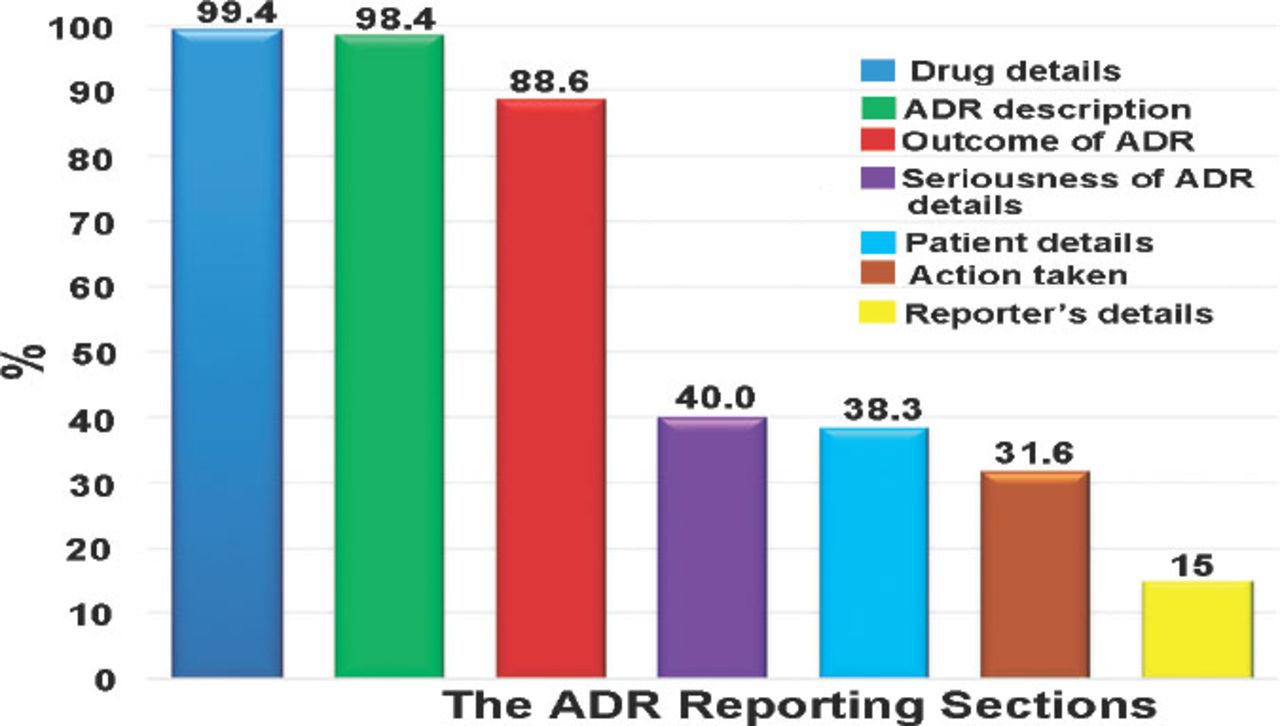
The information related to the drug and adverse was completed in 99.4% and 98.4% of the reports. Assessing the completeness of drug and adverse event fields was carried out separately (namely, not linked). The patient demographic data were filled out in 38.3% of reports, whereas the event outcome information was available in 88.6% of the reports (Figure 1).
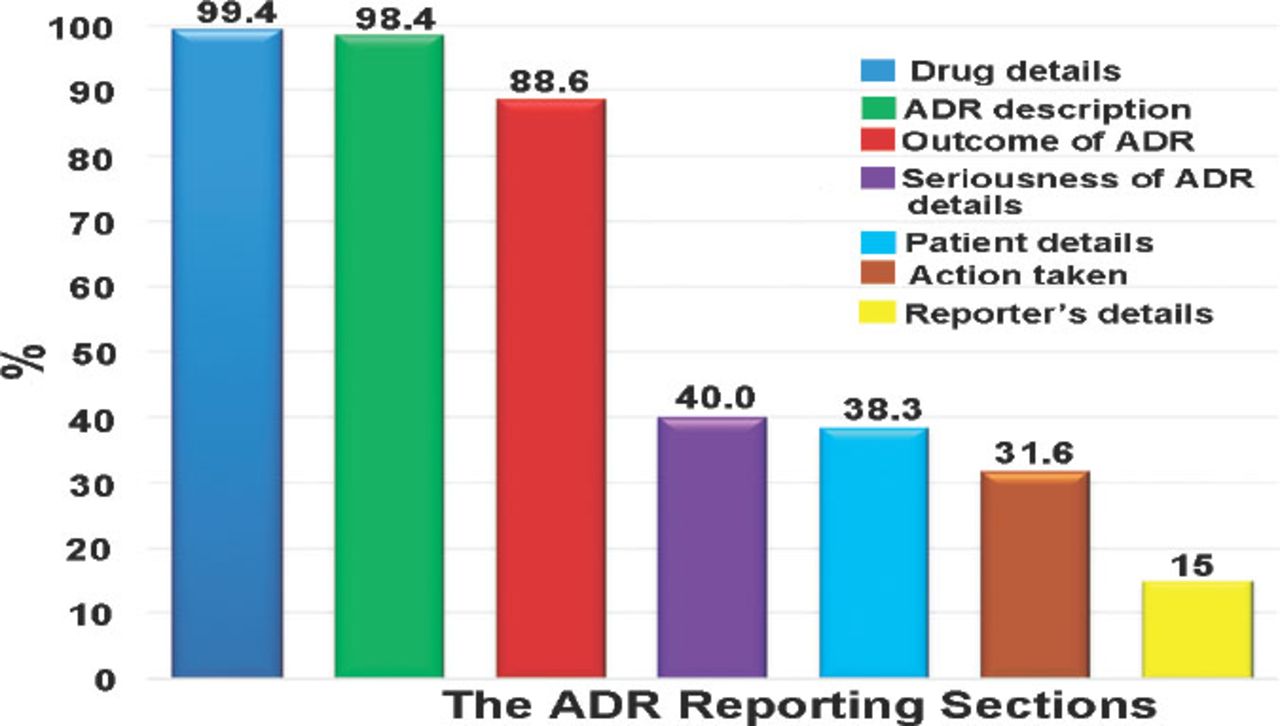
The total report section completeness percentages of the information detailed in reporting forms during the study period. ADR - adverse drug reactions
The gender was completed in 13,356 (90.3%) reports. Furthermore, the age of the patients was completed in 8,712 (58.9%) reports and in 3,862 (26.1%) reports, ‘date of birth’ was completed. We had to create a variable that combined the age and the date of birth fields and the total number of completed reports was 12,574 (85%). Additionally, height was completed in 4,184 (28.3%) reports, and weight in 3,115 (21.1%), while the ‘health institution’ field was completed in only 3,811 (25.8%) reports. The reports in the study included 48,417 drugs. Some drugs occurred in more than one report. The mean was 3 drugs per report, with a range of 1-22 drug(s) per report. In addition, 14,257 (96.4%) reports contained the drugs’ name; 67.2% of these drugs were suspected to have caused the events, and 32.4% of these drugs were concomitant drugs.
Of the 14,545 (98.4%) reports that had event(s), there were 44,787 events mentioned in these reports (that is; some reports have several events in one report). As with the drugs field, the events could occur in more than one report. The mean was 3 events per report and the range was between 1-61 event(s) per report. The dates when the events started (67.2%) and disappeared (19.6%) were completed. Laboratory tests were ordered in 54% of the reports; of those requested laboratory tests, 97.5% of them were completed. In the action taken section of the report, the most reported fields, which were filled out were the drug withdrawn (8.4%) and unknown fields (12%) (Table 1).
Completeness percentage of the information completed in the action taken section of the adverse drug reactions (ADRs).
The ‘event outcome’ was provided in 88.6% of the reports; 9.6% of the events were fatal, 11.2% were not recovered, and 27.8% were recovered. While 9.1% of the events were recovering, 41.6% of the event outcome was unknown. The ‘recovered date’ was provided in 13.7% of the recovered events. One of the most challenging fields was the ‘de-challenge and re-challenge’ section. Only 464 (3.1%) of the reports had the de-challenge field filled in. For the re-challenge field, 17.4% were filled out (Table 2). The seriousness of the event was only filled out in 5,920 (40%) reports. Most of the events (4,877, 82.4%) in the reports were serious. Detail on the type of event seriousness was not available in all the reports. The profession of the person completing the report was the only accessed field in the reporter information section due to the reporters’ confidentiality. However, only 15% of the reports filled up the professions.
Completeness of the information filled-in regarding the outcome of adverse drug reactions (ADRs) section among 14,783 reports received by the pharmacovigilance center.
Assessing the quality of SAERS
Thirty random reports were selected by simple sampling from the SAERS database to assess the quality of information describing the adverse events. Based on the aforementioned criteria in the methods section, the SAERS reports were of good quality and the required information regarding the adverse event was mentioned, was also of good quality.
Perform descriptive statistics for SAERS elements
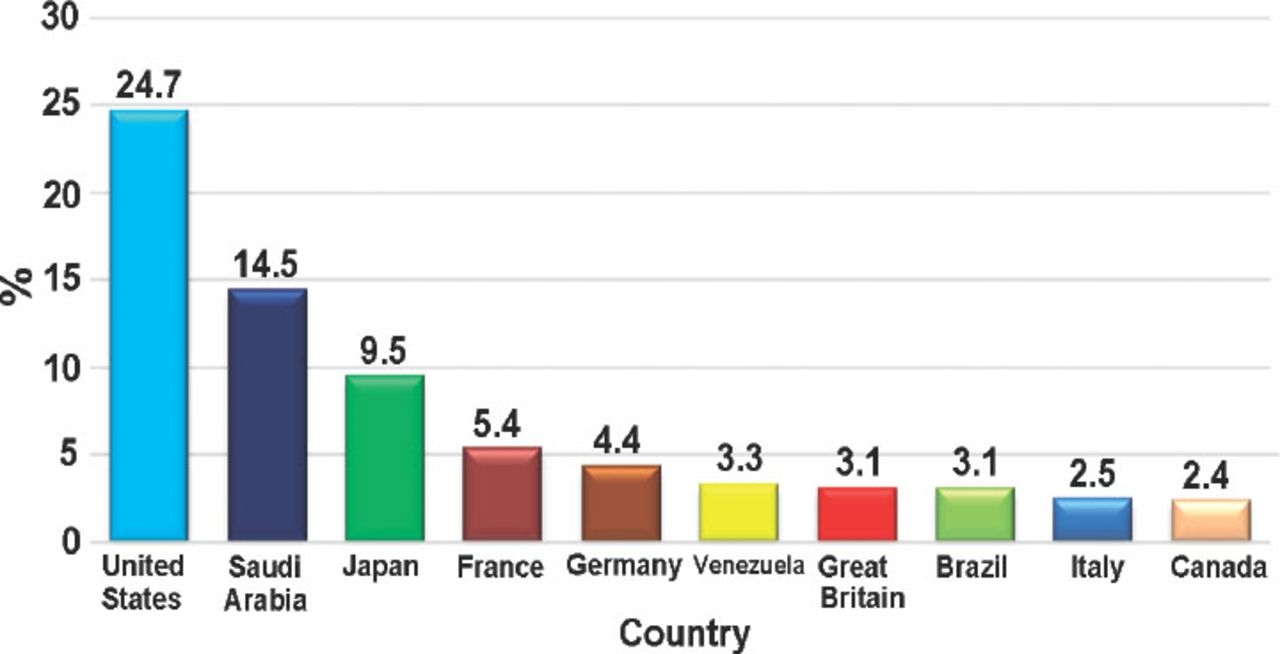
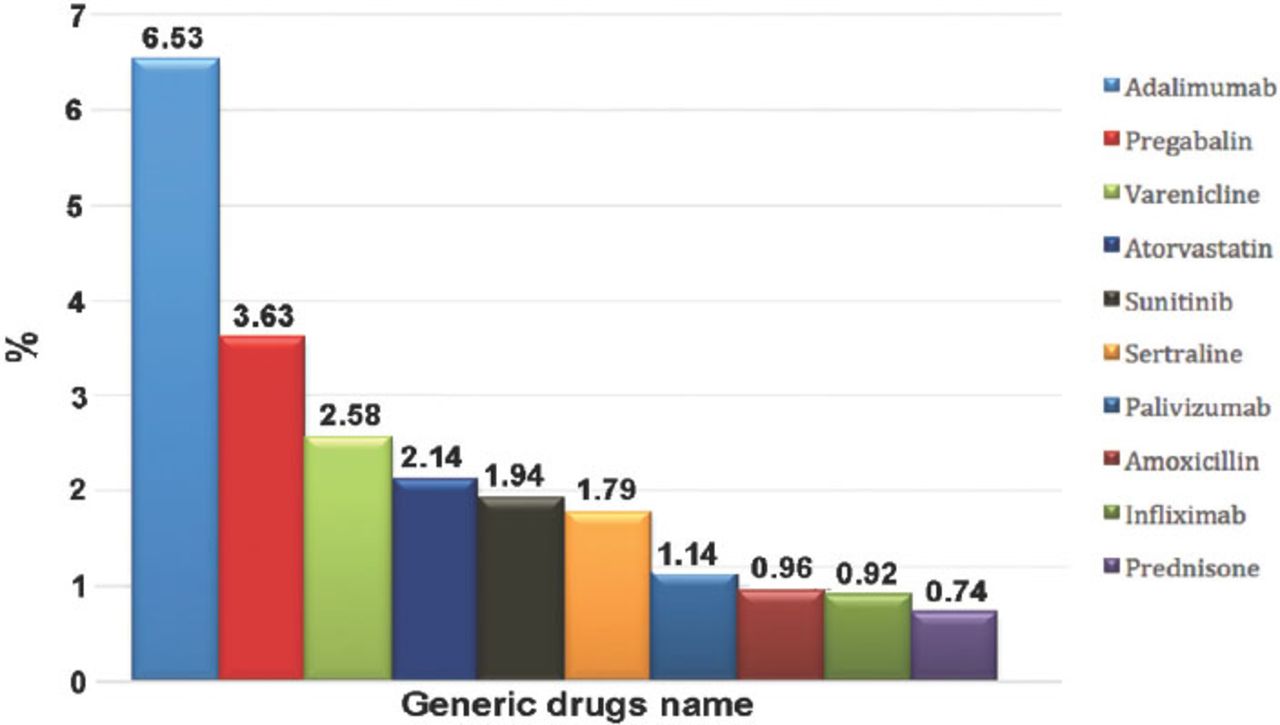
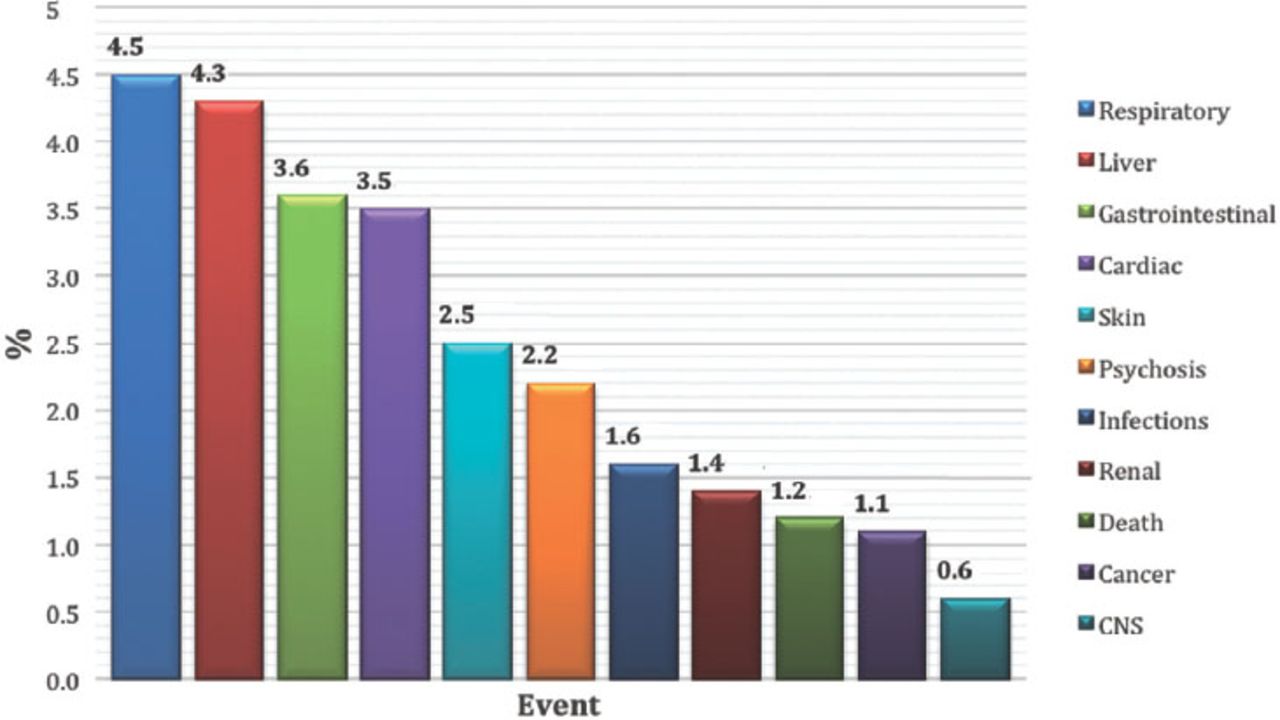
Most of the patients were female; there were 7,364 (55.1%) females compared with 5,992 (44.9%) males. As expected, reported ADRs included more elderly people; as 5,888 (55.8%) of the reports were on patients aged above 60 years old, while only 1,436 (13.6%) reports were on patients aged 20 years old or younger. When the ages were stratified by 10-year intervals, patients between the ages of 60 and 70 years obtained the highest percentage of events reported (18.03%). Report type was provided in 14,545 (98.4%) reports. Eighty percent of the report types were ‘spontaneous’, while 2,827 (19.4%) reports were ‘report from study’, and these were from pharmaceutical companies. Sixty (0.4%) reports were recorded under the type of ‘other’ and 19 (0.1%) ‘not available’. Eighty-five percent of the reports were from international companies, while only 15% of the reports were from Saudi institutions. The reports from pharmaceutical companies were 87.3%. On the other hand, from the healthcare professionals (HCPs), 12.6% of reports were from pharmacists, 0.04% from nurses, and 0.02% of reports were from both physicians and the public. More than 90 international companies reported to the SPS; 24.7% of the companies’ reports were from the United States (Figure 2). Of the reported drugs, 67.2% were suspected to cause the event(s), while 32.4% were concomitant. Only 0.4% of the reported drugs were not known to be suspected or concomitant. The most frequent suspected drugs over the study period were Adalimumab (n=3,695 reports), Pregabalin (n=2,056), Varenicline (n=1,456), and Atorvastatin (n=1,208) (Figure 3). When stratified by year, the most suspected drug in 2009 was Calcium Levofolinate (7.2%, 159), while in 2010 it was Amoxicillin (11.3%, 426). Adalimumab was the most suspected drug in 2011 (n=673, 7.5%), and 2012 (n=1283, 6.8%). The respiratory organ system was the most reported event (4.5%), followed by events related to liver disease (4.3%) (Figure 4).

The most frequently reported adverse events based on country source.

The most frequently reported drugs to Saudi Adverse Event Reporting System between December 2009 and June 2012.

The most frequently reported events among 14,783 reports between December 2009 and June 2012.
Discussion
On March 2009, the SFDA launched the SAERS as a new pharmacovigilance system for monitoring the post-marketing safety of medications.2 Few studies7-9 have assessed the quality and quantity of adverse event reporting systems in different countries. The number of reports to SAERS has doubled each year. In 2009, there were 6% reports; increased to 11% in 2010, 25% in 2011, and 57% in 2012. This shows a good progression in the rate of reporting to SAERS. The highest reported rates to the SPS was from the companies, as it is mandatory by the SFDA that each company should report ADRs against their own drugs. This number was followed by pharmacists, including pharmacist coordinators in local hospitals. Pharmacy coordinators are employees of the hospitals, but they also cooperate with the SFDA to facilitate the reporting methods inside their institutions, and as an access between the hospitals and the SFDA. While reporting from the other HCPs and the public was low, Aggard et al8 (Danish study) found more reports came from physicians (75%) compared with other HCPs (13%), and consumers (11%). Another study by Thiessard et al9 (French study) found similar results from a Danish study. Approximately 91% of reports were reported by physicians and 5% by pharmacists. We speculate that physicians received more training and education with respect to reporting ADRs, and most patients explained their case in detail. Increasing awareness for the HCPs and the public on the SPS, and/or encouraging of reporting is recommended due to the low number of local reports. The spontaneous reporting of adverse events was the most reported type, as it is the more efficient method to identify new ADRs after approval.10,11 The number of adverse events increased with age. In fact, this tendency might be due to an increase in the number of comorbidities and, consequently, an increase in the number of medications used, as those patients become older. Therefore, patients with poly pharmacy would be more susceptible to develop ADRs due to several reasons such as drug-to-drug interaction, drug-food interaction, or drug disease interaction.12-15 Patients between the ages of 60-70 years had the highest percentages of events reported. Our results were similar to those of the French study9 with respect to patients’ age as events occurred more in elderly patients. In addition, our results are similar to Wester et al’s16 study which found that the oldest patients were between 61 and 80 years. Furthermore, female gender was reported more frequently compared with male gender. This distribution was similar to the French study9 and the Swedish study17 since females occurred more in the reports than males. However, our results were different to those reported by Wester et al16 as 57.6% of the reports affected males.9,17 The SAERS presented good quality of information related to the drug and the event, while the information related to patient demographic data and action taken after the event were of a lower quality. Furthermore, the SAERS reports show a good quality related to the adequacy of information describing the adverse event. However, the method of assessing the quality of these reports is not well-defined and there is no global standard for it. Therefore, there is a need for a well-designed structure in evaluating the quality of such reports. In contrast to the SAERS, the Adverse Drug Events Spontaneous Triggered Event Reporting (ASTER) pilot study18 has 100% completed information related to patient demographics. However, the ASTER study has more advantages since the data were received from a triggered electronic health record, which usually contains most of the required data in detail. In addition, our results were similar to a study by Alshammari et al7 who investigated the quality of the Food and Drug Administration (FDA) SAER system and found that the FDA database is of good quality. Nevertheless, it is recommended that awareness is increased on the importance of completing all the patient demographic information to help understand the roles of age, gender, weight, and other information in the occurrence of a specific event. The detailed information related to the reporter was completed least, and only the type of reporter was obtained. This was due to limited access to SAERS due to confidentiality issues. Most reported events were serious, though the seriousness of the types of the events was not clearly described. The French study9 revealed that approximately 44% of the events were serious, which is lower than our results. This could be due to the French database9 having more reports from HCPs who usually reported all types of events (serious and non-serious). A Danish study8 evaluated the reports submitted to the Danish reporting system and found that approximately 52% of the reports were serious. However, the Danish study also evaluated the reports from HCPs and consumers. Similarly, a review of literature on ADRs that occur in children found that on average approximately 26% of the events were serious. Another study19 found that 29% of the reports were serious. While in the case of SEARS, most of the reports were from pharmaceutical companies and regulations state that only serious reports were reported to the SFDA.8,9,17,19 The most reported drug classes were tumor necrosis factor (TNF) inhibitor agents, followed by anticonvulsant agents, smoking cessation agents, and lipid lowering agents. Our results were close to the study of Wu et al20 from England, which found that systemic agents (immunomodulator and antineoplastic) were the most reported drugs followed by analgesic agents and cardiovascular drugs. The French study9 shows that ADRs were most frequently related to nervous system drugs, followed by cardiovascular drugs, and systemic anti-infectives. Wester et al16 found that antithrombotic agents were the top drugs reported in their study followed by diclofenac and antineoplastic agents. However, they investigated the reports associated with suspected fatal events. A literature review19 was carried out on studies assessing the ADRs in children that found that the most reported drug class was anti-infectives for systemic use, followed by antineoplastic and immunomodulating agents, nervous system medications, and respiratory system medications.
Respiratory disorders were the most reported events, followed by events related to liver diseases. Our study result was similar to the study by Davis et al,21 as the most reported events were associated with the respiratory ward. Wu et al20 found that the most reported events were mental and behavioral disorders, cardiovascular consequences, and complications due to injection. Furthermore, the Swedish study17 found that skin disorders were the most reported event in adults followed by neurological disorders, gastrointestinal diseases, and psychiatry and cardiovascular disorders. The study was limited to investigating if any medication error had been made, and that due to the limited access to some information (such as the doses, frequency, route of administration, and indication of what medication was used). In addition, the reporter information was not fully accessed and only the profession type was used due to confidentiality reasons. This study has advantages, as it is the first study to assess the completeness and quality of SAERS.
Implications and recommendations
The information in the database is starting to increase when we compare the starting years and the last year in the study and this could help in the future to assess the signal between a specific event with a specific drug; however, we should keep in mind that some drugs and/or events would have few reports. It is recommended that the report date be added to the reporting forms, which will be helpful for further assessment and evaluation. There are some considerations that the pharmacovigilance department at the SFDA should consider to enhance the data quality by avoiding the use of manual drug entry to prevent spelling mistakes and false duplicated drug frequencies, as well as to stick to the drug name codes and to add the uncoded drug names to the system.
In conclusion, although the SAERS is considered new, it has a high number of reports. Further efforts are required to improve the completeness of the SAERS as a good source to assess the signals between events and suspected drugs especially when it has many reports. In addition, this study found that this database contains relatively good quality data. Furthermore, continuing evaluation of the SAERS’ completeness and quality is recommended.
Ethical Consent
All manuscripts reporting the results of experimental investigations involving human subjects should include a statement confirming that informed consent was obtained from each subject or subject’s guardian, after receiving approval of the experimental protocol by a local human ethics committee, or institutional review board. When reporting experiments on animals, authors should indicate whether the institutional and national guide for the care and use of laboratory animals was followed.
Acknowledgment
The authors would like to thank the vigilance and crisis executive directorate at the Saudi Food & Drug Authority, Riyadh, Saudi Arabia for their help in supplying the information.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received March 18, 2015.
- Accepted May 4, 2015.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.




{kind=link}
{kind=link}
{kind=link}
{kind=link}