


Abstract
Objectives: To investigate the prevalence and significance of different endocrinopathies in children and adolescents with transfusion-dependent thalassemia and sickle-cell anemia.
Methods: This is a descriptive, retrospective study between January 2010 and July 2018 in King Abdulaziz University Hospital, Jeddah, Saudi Arabia. Data was collected through reviewing electronic hospital medical records then filling out data collection sheets and was interpreted through the IBM SPSS Statistics for Windows version 20.0 (IBM Corp, Armonk, NY, USA).
Results: The total sample size was 119 patients, gender equality was almost achieved with 55.5% being male and 45.5% being female. The most common endocrinopathies were identified in the following order of short stature (39.5%), diabetes mellitus (29.4%), hypogonadism (12.6%), osteopenia (12.6%), osteoporosis (9.2%), hypothyroidism (9.2%), hypocortisolism (3.4%), and hypoparathyroidism (2.5%). All of which were statistically significant in their relationship to hemoglobinopathies with the exception of osteopenia and osteoporosis. Hypogonadism and hypocortisolism were found to be statistically significant in their relationship to a positive history of splenectomy at p=0.026 and p=0.012. Short stature was found to be statistically significant in its relationship to the male gender with a p=0.001.
Conclusion: Endocrinopathy is a frequent complication of hemoglobinopathies, for which the most common were found to be short stature, diabetes mellitus, and low bone mineral density.
Hemoglobinopathies are the world’s most common genetically inherited diseases. Thalassemia is recognized as the world’s most common genetic disorder with the level of endocrinopathy experienced by this group being the most significant factor contributing to the quality of life.1,2 Literature shows that more than 80% of those living with transfusion-dependent thalassemia suffer a minimum of one endocrine complication.3 This may be either one of a reduced bone mineral density secondary to thalassemic bone marrow expansion, growth retardation and short stature, hypothyroidism, hypogonadism, hypocortisolism, hypoparathyroidism, or diabetes mellitus.2 The pathophysiology underlying these complications is attributed to the chronic anemia, hypoxia, and iron overload that is constantly developing in these patients.4 The mainstay of management of thalassemic children is through regular blood transfusions with the aim of sustaining a hemoglobin level that is >10 mg/dL. While this approach had dramatically improved the prognosis accompanying thalassemia major, it had also put patients at risk of iron overload and therefore it must be balanced with aggressive chelation therapy to combat iron deposition in the hypothalamic-pituitary axis and endocrine glands.5 It is particularly important for clinicians to recognize, anticipate, and screen for possible endocrinopathies that may complicate cases of severe hemoglobinopathies as in some situations it may be reversible if addressed in a timely manner. This has been demonstrated in patients with biochemical diabetes who once have progressed into clinically overt diabetes, were unable to reverse the condition when compared to those presenting during an asymptomatic phase.6 The aim of this study was to investigate the prevalence and significance of different endocrinopathies in children and adolescents with transfusion-dependent thalassemia and sickle-cell anemia.
Methods
This is a descriptive, retrospective study covering records from January 2010 to July 2018 in Jeddah, Saudi Arabia. Individual patient consent and an ethical approval was obtained from the Research Ethical Committee at King Abdulaziz University prior to the commencement of this study. Written informed consents were obtained from the parents or guardians of the children. All procedure were performed according to the principles of the Helsinki Declaration. Data was collected through reviewing electronic hospital medical records and filling out a data collection sheet. Literature search for similar research was through typing relative MeSH terminology in the PubMed search engine while filtering search results according to publication date, narrowing the results to articles that were published within the past 5 years in order to obtain recent references.
The inclusion criteria were all children aged 18 years or younger with severe thalassemia that is transfusion-dependent or sickle-cell anemia on hypertransfusion regimen and ferritin levels above 600 ng/mL. Subjects were excluded if they expressed features of autoimmunity; had an autoimmune disease such as insulin dependent diabetes mellitus, autoimmune thyroiditis, Addison’s disease, or polyglandular autoimmune endocrinopathy; were diagnosed with idiopathic or familial short stature; had mild or mild-moderate thalassemia or sickle anemia with infrequent blood transfusions, or if their records contained insufficient data. Data collected included demographics such as age, gender, and nationality; anthropometric measures such as height, weight and body mass index; previous history of a diagnosed hemoglobinopathy and splenectomy; age of splenectomy if applicable; frequency of blood transfusion and age in which transfusion treatment was started; type of chelation therapy; biochemical values such as ferritin level and a full endocrine profile including thyroid function, insulin growth factor, fasting plasma glucose, HbA1c, follicle stimulating hormone, luteinizing hormone, oestrogen and testosterone, adrenocorticotropic hormone (ACTH), cortisol, parathyroid hormone, calcium, phosphate, vitamin D, and results of radiological investigations such as a bone mineral density scan. Reference ranges and cut-off values marking the diagnostic criteria for each biochemical test is summarized in Table 1. For those with low basal cortisol or high serum ACTH, an additional cosynotropin test was performed in order to confirm the diagnosis of adrenal insufficiency.
Definition and diagnostic criteria of endocrinopathies used in the present study.
Statistical analysis
Data entry and analysis was conducted using the Statistical Package for Social Sciences, version 24 (IBM Corp, Armonk, NY, USA). Categorical variables were expressed either in frequency whereas continuous variables were expressed in range, mean and standard deviation. Additional tests used included the Chi-square test and t-test, both of which were used to determine the presence or absence of a significant association, with calculated p<0.05 interpreted to be significant.
Results
The total sample size included 119 patients, of whom 55.5% were male and 44.5% were female. The mean age in years was 11.82±5.35. The majority of the sample (87.4%) were diagnosed with transfusion-dependent anemia whereas the remaining 12.6% were diagnosed with severe sickle-cell anemia that required frequent blood transfusions. The average mean for the hemoglobin levels prior to receiving transfusion therapy was 7.89±1.28. The average mean of frequency of blood transfusions per year was around 12.11±3.68 and only 17.6% of the patients had required a splenectomy, which was performed at the mean age of 8.7±5.01. The majority of patients (85.7%) received chelation therapy either orally or through an intravenous infusion. Endocrine complications screened for were osteopenia or osteoporosis, hypothyroidism, hypogonadism, hypocortisolism, diabetes, short stature, and hypoparathyroidism. It was found that 91.6% of the total sample size had at least one endocrine complication with 61.3% having 2 or more endocrinopathies (Table 2). Mean value for ferritin levels during the past year was 3768.77±8636.06 ng/mL. The most common complications were in the following order: short stature (39.5%), diabetes mellitus (29.4%), hypogonadism (12.6%), osteopenia (12.6%), osteoporosis (9.2%), hypothyroidism (9.2%), hypocortisolism (3.4%), and hypoparathyroidism (2.5%) (Figure 1). Of those with hypogonadism, all but one patient had suffered a central cause. Endocrinopathies that were found to be statistically significant in their relationship with a hemoglobinopathy were vitamin D deficiency at a p=0.0001, short stature at a p=0.033, diabetes mellitus at a p=0.013, hypogonadism at a p=0.0001, hypothyroidism at a p=0.0001, and hypocortisolism at a p=0.001. Endocrinopathies that were found to be statistically significant in their relationship to splenectomy were hypogonadism and hypocortisolism at p=0.026 and p=0.012. Endocrinopathies that were found to be statistically significant in their relationship with gender were limited to short stature only at a p=0.001 in which it was more predominant in the male gender. Vitamin D deficiency was found to be an independent association with a prevalence of 73.9%.
Number of endocrine glands affected.
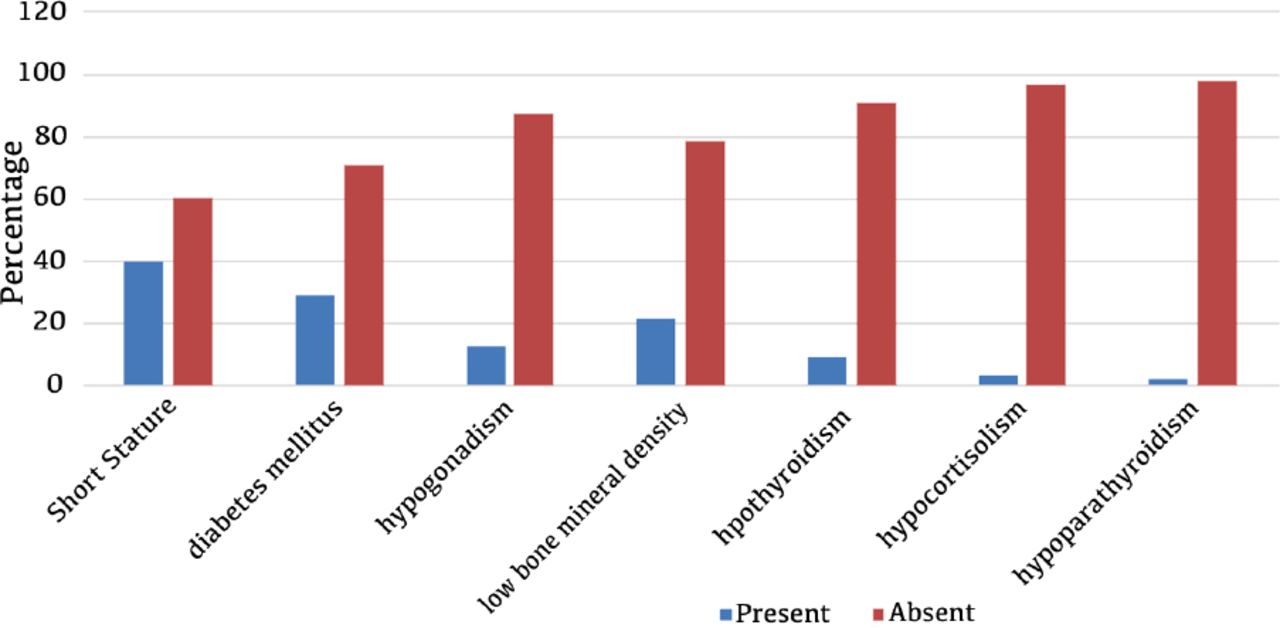
Endocrinopathies complicating transfusion-dependent hemoglobinopathies.
Discussion
Hemosiderosis is a recognized cause for endocrinopathy in patients with hemoglobinopathies as a result of direct organ infilteration, iron deposition, free radical formation, and lipid peroxidation causing mitochondrial, lysosomal and sacrolemmal damage.7 The level of hemosiderosis is monitored through obtaining a serum ferritin level as it is a direct representation of the iron concentration in the blood. Complications are often encountered with serum values that exceed 2000 ng/mL.8 In this study, all children with serum ferritin values that were normal or mildly elevated (less than 600 ng/mL) were eliminated, with the average mean for serum ferritin being 3768.77±8636.06 ng/mL. Short stature and hypogonadism are some of the most common endocrinopathies encountered with hemosiderosis and are more often seen corresponding with ferritin levels that exceed 2500 ng/mL. The hypophysis is particularly susceptible to iron deposition and is therefore one of the most vulnerable organs in states of iron overload.2 Signs of pituitary dysfunction as a result of iron deposition can be manifested through central hypogonadism and hypocortisolism, primary hypoparathyroidism, growth hormone deficiency, or secondary hypothyroidism. It is important to note that the pituitary’s sensitivity is variant from one axis to another, as for example the reproductive axis is affected at a much earlier stage than the thyroid axis. Therefore, many of the cases complicated with hypogonadism are hypogonadotropic in origin, whereas the developing hypothyroidism at first is often a result of direct glandular infiltration, organ siderosis, and chronic tissue hypoxia.9 However, with the progression of hemosiderosis, the hypothyroidism is then gradually caused by a combination of both primary and secondary etiology.
A similar study investigating the prevalence of endocrine disorders in patients with transfusion-dependent thalassemia showed that the most common endocrinopathy encountered was a delayed puberty at a prevalence of 68%, followed by low bone mineral density in 56.1% and short stature in 52.5% of the total sample size.10 The results are somewhat similar to the findings of our study, in which short stature was the most common complication but with a prevalence of 39.5%, though this was succeeded by the development of diabetes mellitus in 29.4% of all sample size. Furthermore, gender was considered as a risk factor for the development of short stature as males were found to be more predisposed than females (p=0.001). Previously, diabetes was thought to occur as a result of direct organ infiltration and iron deposition that destroys the function of pancreatic beta cells. More recently, it is now believed that the development of diabetes in patients with hemoglobinopathies complicated by hemosiderosis is preceded by a prolonged period of insulin resistance that eventually predisposes to the onset of diabete.11,12 The reported prevalence of diabetes in thalassemic patients alone ranges between 4-26%, the upper limit of which is slightly lower than the one identified in this study.6
An overall low bone mineral density was also found in 21.8% of our total sample with 9.2% being osteoporotic. Low bone mineral density in children with hemoglobinopathies is a multifactorial endocrinopathy; for the most part, it is caused by bone marrow expansion secondary to the chronic anemia in suboptimally transfused patients. On the other hand, further contribution to the osteopenia may be a result of either one of the delayed puberty and hypogonadism, hypocalcaemia and hypoparathyroidism, insulin growth factor deficiency and direct iron deposition seen in this patient group.7,13
One study reported a prevalence of 86.8% of their total sample size of transfusion-dependent thalassemics having at least one endocrinopathy,14 which is close to the 87.4% found in our sample and in keeping with the previously published literature.15 The most common endocrine complication encountered was low bone mineral density at a prevalence of 72.6% with a statistically significant bias towards the female gender. This was followed by hypogonadism at a prevalence of 44.5% with a statistical significance correlated to patients who underwent a splenectomy. In addition to hypogonadism, splenectomized patients had approximately double the risk of developing both diabetes and hypoparathyroidism as a complication.14 Although more known for its role in overwhelming infectious complications, splenectomies were also investigated as a separate risk factor for the development of endocrinopathies in patients with thalassemia.16,17 In this study, a positive history of a splenectomy was considered as a statistically significant risk factor for the development of hypogonadism p=0.026 and hypocortisolism at p=0.026. This might be explained by the role splenectomies play in rapidly precipitating an increase in circulating ferritin levels and favoring the deposition of iron in multiple organs through decreasing the body’s total iron binding capacity.18
Another study showed that the overall prevalence of endocrine complications in post-splenectomy patients is twice the overall prevalence of endocrinopathies seen in non-splenectomised patients, with 86.7% of those who had a splenectomy suffering a complication of at least one endocrinopathy compared to the 43.7% seen in those without a splenectomy.19
Hypoparathyroidism as a complication of transfusion-dependent hemoglobinopathies was reported by some studied in prevalences of 9.4 %and 13.2% whereas others reported a prevalence of 0%.10,14,19 In published literature, it has a more frequent prevalence between 3.6% to 22.5%; however, in the present study it was considered the least frequent endocrinopathy with an overall prevalence of only 2.5%.20,21
In addition, vitamin D deficiency was studied as an association in patients with severe hemoglobinopathies. It was found that 73.9% of the total sample had suffered a vitamin D deficiency. Previous reports have provided prevalences ranging from 12% up to a total of 100%of the total sample suffering from an associated vitamin D deficiency.22,23 This could be a result of nutritional deficiency but may also be explained by an impaired 25-hydroxylation due to iron deposition in the liver.24
In conclusion, endocrinopathy is a frequent complication of hemoglobinopathies, for which the most common were found to be short stature, diabetes mellitus, and low bone mineral density. For prevention, we recommend that children and adolescents with transfusion-dependent hemoglobinopathy should be managed with aggressive chelation therapy and be followed up regularly for early screening of endocrinopathies.
Acknowledgment
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant No. (DF-135-140-1441). The authors, therefore, gratefully acknowledge DSR technical and financial support.
Footnotes
Disclosure. This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant No. (DF-135-140-1441).
- Received September 29, 2019.
- Accepted December 12, 2019.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.




{kind=link}