


ABSTRACT
Acute myeloid leukemia (AML) is an aggressive leukemic malignancy that affects myeloid lineage progenitors. Relapsed or refractory AML patients continue to have poor prognoses, necessitating the development of novel therapy alternatives. Adoptive T-cell therapy with chimeric antigen receptors (CARs) is an intriguing possibility in the field of leukemia treatment. Chimeric antigen receptor T-cell therapy is now being tested in clinical trials (mostly in phase I and phase II) using AML targets including CD33, CD123, and CLL-1. Preliminary data showed promising results. However, due to the cellular and molecular heterogeneity of AML and the co-expression of some AML targets on hematopoietic stem cells, these clinical investigations have shown substantial “on-target off-tumor” toxicities, indicating that more research is required. In this review, the latest significant breakthroughs in AML CAR T cell therapy are presented. Furthermore, the limitations of CAR T-cell technology and future directions to overcome these challenges are discussed.
Introduction
Acute myeloid leukemia (AML) is the fast-advancing destructive neoplasm of myeloid precursors that causes partial/complete differentiation block and results in bone marrow (BM) failure.1 Acute myeloid leukemia is characterized by genomic mutations and chromosomal abnormalities within BM cells, interferes with natural cell processes, and leads to uncontrolled cell expansion and compromised differentiation. It represents approximately 23% of all leukemia cases.2 Although AML is common in adults, the incidence estimates in the United States are 7.8 per million individuals aged 0-14 and 9.1 per million individuals aged 15-19 years.3 In 2023, the American Cancer Society estimated 20,380 new cases of AML, with approximately 11,310 cases becoming deadly and an average age of diagnosis of ~68 years.4
Acute myeloid leukemia results from 2 or more mutations, one of which promotes proliferation (class I mutations) while the other inhibits differentiation (class II mutations).5 FLT3-ITD, KRAS, and KIT mutations are classified as class I, while mutations involving core binding factor (CBF) fusions and CEBPA are classified as class II.5 Approximately half of individuals suffering from AML display a normal karyotype and mutations in certain genes, such as FLT3, IDH1, IDH2, and NPM1, while the remaining exhibit chromosomal abnormalities affecting chromosomes 5, 7, 9, 16, 8, 21, 15, or 17.6
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a conventional treatment for AML. Meanwhile, targeted therapies using inhibitors of FLT3, IDH1, IDH2, BCL2, and others and antibody-based therapies that employ anti-CD33 and anti-CD3 have emerged as novel AML treatments.7-11 However, treatment resistance and relapse are still major challenges, emphasizing the pressing demand for innovative therapeutic strategies.12
An exciting immunotherapeutic strategy that has revolutionized the field of blood cancer treatment involves modifying autologous/allogenic T-cells using chimeric antigen receptors (CARs), enabling the specific targeting of leukemia-associated antigens. This review provides an overview of the various antigens used in CAR T-cell therapy for AML, presents current clinical findings, and discusses the limitations of CAR T-cell technology and future directions to overcome these challenges.
CAR T-cell therapy
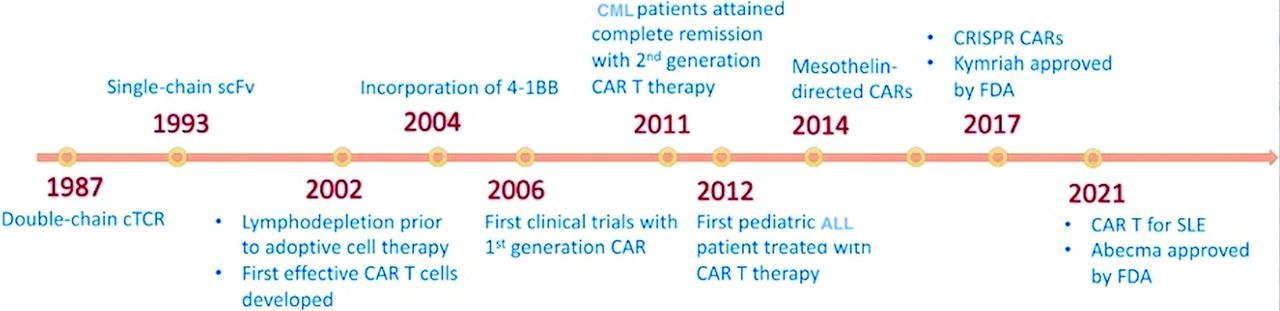
Chimeric antigen receptors are recombinant antigenic determinants designed by uniting the attaching domain of immunoglobulins with that of a signal-transducing domain derived from T-cells.13 By utilizing lentiviral/retroviral vectors, CARs can be genetically expressed on the surface of αβ CD4+ and CD8+ T-cells. This modification directs T-cells toward a specific antigen, initiating a signaling cascade inside CAR-engineered T-cells, resulting in self-renewal and tumor-specific cytotoxic activity similar to that of stimulated T-cells.14 Figure 1 shows a timeline of the major developments in CAR T-cell therapy.

- The timeline of major developments in chimeric antigen receptor T-cell therapy. CML: chronic myeloid leukemia, CAR: chimeric antigen receptor, CRISPR: clustered regularly interspaced short palindromic repeats, FDA: Food and Drug Administration, cTCR: chimeric T-cell receptor, SLE: systemic lupus erythematosus
In 2017, the Food and Drug Administration (FDA) sanctioned tisagenlecleucel as the pioneer CAR T-cell treatment regime for treating pediatric and young adult acute lymphoblastic leukemia (ALL).15 This was followed by the approval of the following additional CD19-based CAR T-cell therapies for various B-cell leukemias such as: axicabtagene ciloleucel, brexucabtagene autoleucel, and lisocabtagene maraleucel.16-18 In subsequent years, the FDA granted approval for 2 B-cell maturation antigen (BCMA)-specific CAR T-cell therapies for treating multiple myeloma (MM) such as: idecabtagene vicleucel and ciltacabtagene autoleucel.19,20
CAR T-cell therapy in AML
In 2013, a noteworthy clinical trial marked a pivotal moment in CAR T-cell therapy for AML. This study involved the first reported use of second-generation anti-CD28-ζ CAR T-cells targeting Lewis Y in AML. Although leukemia progression was observed, this investigation established the tolerability profile of administering CAR T-cells to individuals with AML; however, on-target/off-tumor effects were reported.21
A key step in the development of CAR T-cell therapy in AML involves selecting the appropriate surface antigen, which should have a robust expression on leukemic cells and negligible or low expression on normal cells to avoid on-target/off-tumor effects. Several cell surface antigens, including CD123, CLL-1, CD33, CD44v6, and FLT3, have been identified as possible targets. Table 1 lists the clinical trials for the treatment of AML employing various targets.
- Registered clinical trials using chimeric antigen receptor T-cell for acute myeloid leukemia in https://clinicaltrials.gov/ as of 8/3/2024.
CD33 is a ubiquitous membrane-spanning member of the sialic acid-binding immunoglobulin-like lectin (SIGLEC) group. While it is frequently present in tissue-resident macrophages, normal progenitors, and approximately 90% of blast cells, it is rarely detected on embryonic CD34+ hematopoietic stem cells (HSCs).22,23
CD33 remains one of the most researched drug candidates for AML management. Experiments using second-generation anti-CD33 CAR T-cells in NOD SCID gamma (NSG) mouse models with AML have demonstrated a notable decrease in AML burden and an increase in animal survival.24 Additionally, preclinical outcomes revealed that anti-CD33 CAR T-cells reduced AML blasts and sustained mouse endurance in xenogeneic animal models while causing cytopenia and a decrease in myeloid progenitors.25 Furthermore, a third-generation CAR T-cell directed at CD33 (3G.CAR33-T) demonstrated enhanced viability, secretion of cytokines, and robust cytolytic action when exposed to normal and leukemic CD33+ cells in comparison to second-generation anti-CAR33 CAR T-cells.26
A phase 1/1b study (NCT03927261) investigating PRGN-3006 UltraCAR T-cells, featuring a CD33-specific CAR construct, membrane-bound IL-15 (mbIL15), and a kill switch in patients with relapsed or refractory (R/R) CD33+ AML is ongoing. In a preclinical investigation, 6 different anti-CD33 CAR T-cells incorporating CD3ζ peptide and CD28 or 4-1BB costimulatory domains exhibited notable anticancer action.27 Encouraged by these promising outcomes, a phase I clinical trial (NCT03971799) is currently ongoing to establish the maximum tolerated dose (MTD) of lentivirally transduced autologous and allogenic anti-CD33 CAR T-cells for individuals with R/R AML and post-HSCT R/R AML.27 In an early phase I trial (NCT01864902) for R/R AML, a patient administered approximately 109 autologous T-cells (of which 38% were transduced) exhibited cytokine release syndrome (CRS) and pancytopenia. Despite a noticeable decrease in observed myeloid blast cells 2 weeks after treatment, there was subsequent blast cell rebound, resulting in relapse 9 weeks post-therapy.28 A safety evaluation with varying dosages of autologous CD33-CAR T-cells (NCT03126864) showed adverse events, including CRS and neurotoxicity.29
IL-3 receptor subunit (IL-3Rα, CD123) levels are elevated not only in leukemia stem cells (LSCs) and AML blasts but also in early HSPCs, resulting in the risk of long-lasting or even permanent myeloablation.30 CD123 stimulates cell propagation when exposed to IL-3 and has high expression in AML cells, with a negligible expression on CD34+ BM cells, rendering it a suitable candidate for CAR T-cell therapy in AML.31,32
In preclinical studies, anti-CD123 CAR T-cell therapy effectively eliminated AML blast cells while demonstrating acceptable levels of toxicity to HSPCs.33,34 Furthermore, 4-1BB- and CD28-specific CD123-CAR T-cells proved capable of targeting AML cells while profoundly ablating normal hematopoietic regeneration.35
In an initial phase I trial (NCT02623582), autologous T-cells with anti-CD123 linked to TCR/4-1BB regions were evaluated in 5 patients with R/R AML. The patients underwent lymphodepletion therapy and then received CD123 CAR T-cells. However, due to adverse events, including CRS, a lack of anti-leukemic efficiency, and reported on-target/off-tumor toxicity, the trial was terminated.36 Another ongoing investigation into allogeneic CD123 CAR T-cells was halted due to patient death 9 days after infusion. The trial resumed with conditions including a reduction in the dose of CART123 and chemotherapy, as well as not exceeding the age of 65 years.37
To enhance safety, novel anti-CD123 CAR T-cell therapy, which exploits the rapidly adjustable universal CAR T-cell platform (UniCAR) to improve tolerability while retaining full anti-AML effectiveness, was developed. A dose-escalating clinical study (NCT04230265) is currently underway to investigate the therapeutic advantage of the novel UniCAR system.38 Moreover, demethylating agent-based drugs have shown promise in enhancing immunological reactions and assisting cancer cell eradication by anti-CD123 CAR T-cells.39
Initial findings from the first cohort of an ongoing first-in-human (FIH) investigation using lentiviral-infected CD123-specific CD28-CAR T-cells demonstrated that one of 2 patients who underwent treatment with a dose of 50 million CAR+ T-cells achieved an AML-free status phenotypically for a duration of 2 months (NCT02159495). Shortly after, the individual underwent a second HSCT therapy, which resulted in a blast cell decrease from 77.9% to 0.9%.40 Furthermore, among the 4 subjects tested, one individual achieved complete remission (CR) with a higher dose of 200 million CAR T-cells, another with CR prior to the start of the therapy continued to have CR, and the remaining 2 subjects experienced a decrease in the number of AML blasts with no relapse. Notably, all toxicities were reversible and tolerable, with no known dose-limiting toxicities or concomitant cytopenia.41
The natural killer (NK) group 2D (NKG2D) receptor in AML exhibits selective expression on monoblasts, while myeloblasts and chemoresistant LSCs either lack or show weak expression, which enables them to evade immune defense and prevent NK-mediated apoptosis.42 Hence, utilizing CAR constructs to activate NKG2D ligands represents a valuable strategy for immunotherapy in AML.43 As such, a bispecific FLT3-CAR therapy incorporating scFv and NKG2D-CAR T-cells has been developed, demonstrating effective eradication of AML blasts in preclinical studies.44
CYAD-01 are recombinant autologous T-cells expressing a CAR engineered by linking the NKG2D target with the CD3ζ signaling component.45 A preliminary phase I FIH study (NCT02203825) revealed encouraging activity of CYAD-01 in subjects with AML.46 Another trial (NCT03018405) is being carried out to assess the safety and tolerability of large doses and repeated injections of NKG2D-CAR T-cells.45,47 Although it showed antileukemic activity, patients experienced adverse events and CRS associated with the 3 dose levels, warranting further investigations into safety and efficiency.45
CLL-1 is highly expressed in approximately 92% of AML blasts and granulocytes, while it is expressed in approximately 2.5% of HSCs, making it a suitable target for CAR T-cell therapy.48,49 Anti-CLL1 CAR T-cell therapy in a child with secondary AML resulted in favorable outcomes, including molecular CR for more than 10 months.50 A phase I/II anti-CLL1 CAR T-cell therapy trial involving 4 children with R/R AML resulted in 3 cases of CR with minimal residual disease (MRD) negativity.51 In a phase 1 anti-CLL1 CAR T-cell therapy trial involving adult R/R AML, most patients experienced CRS, although it was tolerable when patients were followed up for 6 months.52 Treatment with dual-targeting CD33- and CLL-1-specific CAR T-cell therapy in a phase I trial involving a child with complex karyotype AML achieved CR.53 As CLL-1 is highly expressed in granulocytes, it is recommended that anti-CLL1 CAR T-cell therapy be bridged with HSCT to avoid granulocytopenia.
FLT3-ITD and FLT3-TKD aberration is present in ~20% of all AML cases and FLT3-TKD aberration is present in 7% of all AML cases, with only a subset of HSPCs expressing FLT3.54 Nevertheless, tailored CAR technologies are not aimed at mutant FLT3 but rather at FLT3 as a whole.55,56 Second-generation FLT3-specific CAR with a 4-1BB costimulatory domain displayed noteworthy cytolytic effects on FLT3+ AML as well as on primary cell lines, with negligible off-tumor damage to healthy HSCs.57 Some studies have reported blood cell-related toxicity in animal models.56
CD44v6, splicing variant 6 of the hyaluronic acid receptor (CD44, a class I transmembrane glycoprotein), is elevated in AML but not in HSCs and has insignificant expression in normal cells.58,59 A second-lineage CAR redirected toward CD44v6 was developed and showed cytotoxic activity against AML blasts but not HSCs.60 Clinical trials of CD44v6-based CAR T-cell therapy for the management of AML patients (NCT04097301) are currently underway.61
CD7 expression is reported in approximately 30% of AML cases and its absence in HSCs makes it a good candidate for CAR T-cell therapy.62,63 Utilizing CD7-specific CAR T-cell therapy in CD7 knockout T-lymphocytes has been proven to target CD7+ AML cells with no toxic effects on myeloid and erythroid cells.64
CD38 is highly expressed in AML samples and hematopoietic progenitors but is absent in long-term HSCs.65,66 Preclinical studies using CD38-CAR T-cells have shown antileukemic activities in AML cell lines and primary AML associated with improved survival while targeting normal hematopoietic progenitors.67
Limitations of CAR T-cell therapy
Chimeric antigen receptor T-cell therapy faces a formidable challenge when tumor cells downregulate the target CAR antigen. This results in a partial or complete loss of antigen expression and contributes to disease relapse following CAR T-cell therapy.68 This has been observed in ALL and MM patients in which CD19 and BCMA expression, were downregulated or lost after treatment with CAR T-cells.69,70 This phenomenon of antigen escape has also been reported in solid tumors. The expression of IL13Ra2 was downregulated in recurring glioblastoma cases after CAR T-cell therapy.71 To overcome this issue, increase the efficiency of CAR T-cell therapy, and reduce tumor recurrence/relapse rates, strategies involving targeting multiple antigens using dual CAR constructs or tandem CARs have been developed. Data from clinical trials using CD19/CD22 and CD19/BCMA dual-targeted CAR T-cells for ALL and MM, have shown promising results and prolonged remission rates. However, increased on-target off-leukemia toxicity has been associated with dual-target CAR constructs versus single constructs.72,73 In AML, compound constructs have been developed to enhance anti-AML activity. Chimeric antigen receptor T-cell dual-target CD123/NKG2DLs and CAR T-cell dual-target CD33/TIM3 have been shown to be efficacious against AML cell lines and primary AML.74,75 Although the dual-target strategy has proven to be effective against antigen escape, further improvements are needed to overcome other hurdles, such as on-target off-tumor effects.
Targeting AML antigens is challenging due to their frequent expression in normal hematopoietic cells, necessitating careful antigen selection in CAR design to achieve high therapeutic efficiency and avoid on-target off-tumor toxicity.76 As this is a common issue between hematological and solid tumors, an emerging strategy to address this challenge involves directing CARs toward tumor-associated posttranslational modifications (PTMs), such as overexpressed truncated O-glycans in solid tumors.77 A total of 4 primary PTMs directed toward CAR T-cell glycated antigens have been explored.76
Optimizing the affinity of scFvs can help mitigate the on-target off-tumor effect of antigens that are highly expressed in tumor cells but have low expression in normal cells. CD38-CAR T-cells engineered to have a low affinity have been shown to eliminate CD38+ MM cells, while CD38+ hematopoietic cells remain unaffected.78 This strategy may be effective in designing CAR T-cells for AML due to the abundance of common antigens and the variable expression between AML and normal blood cells.
The toxicity associated with CAR T-cell therapy represents an obstacle to achieving the maximum benefit of CAR T-cell therapy as a standard of care therapy. Such toxicities include CRS, hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS), and immune effector cell-associated neurotoxicity syndrome (ICANS).79 Severe and life-threatening events have been reported, even for FDA-approved CD19-targeted CARs.80,81 In CAR T-cell treatment for ALL, nearly all patients experience some toxicity, with 23-46% developing CRS and extensive T-cell proliferation in vivo.82,83 CRS and ICANS become more severe following the infusion of a high number of CAR T-cells and upon the secretion of the cytokines IL-1 and IL-6.84 Further research is required to identify new strategies to overcome the toxicity associated with CAR T-cells.
The magnitude and dynamics of CAR T-cell stimulation are intricately affected by multiple factors, including the extent of cancer antigen expression, the affinity of the antigen-binding domain to its target epitope, and the integration of costimulatory components within the CAR.85 Thus, optimizing therapeutic efficacy and minimizing toxicity require careful consideration of multiple components within the CAR’s modular structure.
Making modifications to the hinge and transmembrane domains of CARs is one strategy to reduce toxicity.86 In a phase 1 trial involving patients with B-cell lymphoma, CD19-targeted CAR T-cells with genetic alterations resulting in a longer CD8a hinge and intracellular domain sequences led to lower cytokine production, reduced CAR T-cell propagation, absence of grade 1 CRS or neurotoxicity in all patients, and CR in 54.5% of patients when compared to shorter CD8a hinge and intracellular domain sequences.87 Additionally, the host’s immune response to CAR constructs may pose a challenge. Thus, using human-derived fragments instead of murine-derived fragments is an optimal solution to avoid immunological-related toxicity.88
Another recent approach to preventing CAR T-cell cytokine toxicity involves the inhibition of granulocyte-macrophage colony-stimulating factor (GM-CSF) production either by modifying CAR-transduced T-cells or by combined treatment. Preclinical studies indicate that mutational inactivation of GM-CSF by generating GM-CSF-deficient CD19-CAR T-cells through the CRISPR/Cas9 disruption of GM-CSF or the inhibition of GM-CSF with lenzilumab decreases neurotoxicity and CRS while enhancing CAR T-cell efficacy.89
Employing “off-switches” or suicidal genes is another strategy to reduce CAR T-cell toxicity. These approaches enable the selective reduction of customized CAR T-cells during adverse reactions by treating them with an additional stimulating agent.90 Various approaches have been developed, including inducible Cas9 switches with immediate effects on eliminating CAR T-cells in approximately 30 minutes, protease-based small molecule-assisted shutoff CARs (SMASh-CARs), and the administration of dasatinib (a tyrosine kinase inhibitor) immediately after CAR T-cell injection to reversibly inhibit CAR T-cell activation, thereby protecting model animals from CRS-induced mortality.91-93 Such strategies would be highly beneficial if they are engineered to be temporally controlled to allow for the inhibition and activation of CAR T-cells when needed.
Conclusion and future directions
Despite advancements in understanding AML etiology and pathophysiology, disease relapse continues to be the primary cause of mortality, even following HSCT. Chimeric antigen receptor-based immunotherapeutic intervention is a promising approach with the potential for long-term protection in individuals with AML relapse. However, the success of CAR T-cell therapy in AML depends on various factors. Overcoming immune escape and identifying AML-specific target antigens remain critical hurdles in successfully instigating adoptive CAR T-cell therapies in the future. Other ongoing considerations include optimizing CAR signaling, managing CAR T-cell persistence post-treatment to avert extended myeloablation, host conditioning complications, and mitigating T-cell-mediated toxic reactions. With numerous clinical trials in progress, assessing the ongoing safety of CAR T-cell therapy and its ability to target relapse-inducing LSCs will be intriguing. There is optimism that therapeutic alternatives will continue to advance, offering improved efficacy with reduced toxicity for better patient outcomes.
Acknowledgment
The author would like to thank the Deanship of Scientific Research at Shaqra University, Shaqra, Saudi Arabia, for supporting the work. The author also would like to thank Scribendi Inc for the English language editing.
Footnotes
Disclosure. Author has no conflict of interests, and the work was not supported or funded by any drug company.
- Copyright: © Saudi Medical Journal
This is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.
- 9.
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.
- 93.↵
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.