


Abstract
Objectives: To identify potential diagnostic markers for small cell lung cancer (SCLC) and investigate the correlation with immune cell infiltration.
Methods: GSE149507 and GSE6044 were used as the training group, while GSE108055 served as validation group A and GSE73160 served as validation group B. Differentially expressed genes (DEGs) were identified and analyzed for functional enrichment. Machine learning (ML) was used to identify candidate diagnostic genes for SCLC. The area under the receiver operating characteristic curves was applied to assess diagnostic efficacy. Immune cell infiltration analyses were carried out.
Results: There were 181 DEGs identified. The gene ontology analysis showed that DEGs were enriched in 455 functional annotations, some of which were associated with immunity. The kyoto encyclopedia of genes and genomes analysis revealed that there were 9 signaling pathways enriched. The disease ontology analysis indicated that DEGs were related to 116 diseases. The gene set enrichment analysis results displayed multiple items closely related to immunity. ZWINT and NRCAM were screened using ML and further validated as diagnostic genes. Significant differences were observed in SCLC with normal lung tissue samples among immune cell infiltration characteristics. Strong associations were found between the diagnostic genes and immune cell infiltration.
Conclusion: This study identified 2 diagnostic genes, ZWINT and NRCAM, that were related to immune cell infiltration by integrating bioinformatics analysis and ML algorithms. These genes could serve as potential diagnostic biomarkers and provide possible molecular targets for immunotherapy in SCLC.
Lung cancer is a prevalent malignancy and a significant contributor to cancer-related mortality on a global scale. The prevalence and fatality rates are high, especially in China.1,2 Based on its biology, therapy, and prognosis, lung cancer comprises 2 main types: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). Approximately 70% of lung cancer cases diagnoses occur at an advanced stage, rendering them inoperable. Distinguishing SCLC from NSCLC begins with morphological analysis supported by immunohistochemistry, followed by molecular techniques.3 Small cell lung cancer is an invasive neuroendocrine carcinoma that accounts for approximately 15% of all lung cancer cases. At the time of diagnosis, more than 70% of SCLC cases have already metastasized. Furthermore, the 5-year survival rate of patients with metastases is less than 1%.4-6 Although SCLC patients initially respond well to first-line treatment, most experience recurrence, and few therapeutic advancements have been carried out over the last 3 decades. Hence, SCLC is considered a recalcitrant cancer.7 Therefore, finding new and probable diagnostic markers is vital for the diagnosis and therapy of SCLC.
Using biotechnology and immunological methods, immunotherapy is a novel modality to boost targeted immune responses against cancers and stimulate the body’s immune system to selectively eliminate cancerous cells. This immunostimulatory ability extends beyond primary tumors and demonstrates a remarkable ability to combat metastatic tumors.8-10 The tumor microenvironment (TME) is primarily composed of stromal cells, immune cells, and extracellular matrix, and changes in these components produce several physiologically distinct and specialized TME, the main cancer immunotherapy ways targeting the immune components of TME contain the use of adoptive-T lymphocytes, CAR-based therapies, cancer vaccines and immune checkpoint inhibitors.11 A substantial body of evidence suggests that intratumor heterogeneity (ITH) along with the interactions within the TME, plays a crucial role in various aspects of tumor biology and therapeutic responses.12 For instance, the diverse states of T-cells in SCLC can offer potential immunotherapeutic targets and indicate that specific patients who respond to immunotherapy have more substantial benefits.13 Due to the diversity of cancers and variations in each individual’s immune system, immunotherapy may not produce favorable treatment outcomes for everyone. In contrast to highly immunogenic cancers, SCLC has fallen behind in the field of immunotherapy in the past decade. However, recent advancements in cancer immunotherapy research offer new hope for patients with SCLC, potentially providing them with better and more sustainable survival opportunities despite numerous unresolved challenges.6,14
Machine learning (ML) is a branch of artificial intelligence that concentrates on employing mathematical algorithms to detect patterns in data for the purpose of making predictions.15 Machine learning-based methods play an important role in integrating and analyzing the extensive and complicated datasets and are increasingly applied in clinical oncology to diagnose cancers, predict patient prognosis, and provide information for treatment plans.16,17 The development of bioinformatics has a long time, with the purpose of utilizing information science and statistical methods to understand biological phenomena.18 It has been widely used for comparative genomic, transcriptomic, and bacterial microbiome analysis in sequencing, animal cell biology, and plant physiology in imaging.19 Therefore, it is particularly important to apply ML and bioinformatics methods to identify diagnostic genes and immune cell infiltration characteristics of SCLC, which providing potential biomarkers for diagnosis of SCLC and searching for possible molecular targets for immunotherapy.
Methods
Datasets GSE149507, GSE108055, GSE73160, and GSE6044 were downloaded from the gene expression omnibus (GEO) database. The GSE149507 dataset was generated using the GPL23270, consisting of 18 SCLC and 18 adjacent lung tissues. The platform for GSE108055 was GPL13376, which included 12 SCLC tissue samples and 10 adjacent normal lung tissue samples. The platform for GSE73160 was GPL11028, which contained most of the SCLC cell lines. The GSE6044 platform was GPL201 and contained 9 SCLC tissues and 5 normal lung tissues. Each dataset was normalized using the normalize between arrays function in the limma R package, and all gene expression data were log2 transformed. The GSE149507 and GSE6044 datasets were merged, and the batch effect was removed to serve as the training group. The GSE108055 served as validation group A, whereas GSE73160 served as validation group B.
The limma package of R served as a filter for differentially expressed genes (DEGs) in SCLC with normal lung tissues among the training group. Genes with a corrected p-value of <0.05 and |log fold change (FC)| >2 were regarded as DEGs. The pheatmap R package was employed to generate the heatmap of DEGs, while the ggplot2 and ggrepel R packages were used for creating the volcano plot.
The functional enrichment analysis of DEGs was carried out using the ggplot2, enrichplot, org.Hs.eg.db, clusterProfiler, and DOSE R packages, which included gene ontology (GO), kyoto encyclopedia of genes and genomes (KEGG), and disease ontology (DO) analyses, with the setting of p-valueFilter=0.05 and q-valueFilter=0.05 (corrected p-value) as filtrating conditions. Additionally, gene set enrichment analysis (GSEA) of functions and pathways between SCLC and normal lung tissues in the training group was carried out using the gene sets c5.go.v7.4. symbols.gmt, and c2.cp.kegg.v7.4. symbols.gmt.
Least absolute shrinkage and selection operator (LASSO) and support vector machine-recursive feature elimination (SVM-RFE) methods were applied for identifying candidate diagnostic genes from DEGs. The LASSO algorithm is recognized as a compressive estimation model that can eliminate insignificant variables by implementing a penalty function, thereby compelling the compression of multiple regression coefficients. Serving the maximum interval principle of support vector machines as base, the SVM-RFE algorithm is a sequential backward selection method which adheres to the principle of structural risk minimization while also aiming to minimize empirical errors. The LASSO model was constructed by the use of the glmnet R package. Genes corresponding to this point were selected, with the minimum cross-validation error. The e1071, kernlab, and caret packages in R were applied to construct the SVM-RFE algorithm. The intersecting genes identified using the Venn R package were considered candidate diagnostic genes.
In validation group A, the ggpubr R package was applied for validating the variance in expression of candidate diagnostic genes in SCLC with normal lung tissues. Receiver operating characteristic (ROC) curves were employed to evaluate the predictive effectiveness of the candidate genes in both the training group and validation group A. Furthermore, the differential expression of potential biomarkers was analyzed between 64 SCLC and 2 normal lung cell lines in validation group B. The stat_compare_means function was used for the statistical analysis.
The expression of candidate diagnostic genes was further verified using quantitative real-time polymerase chain reaction (qRT-PCR) in BEAS-2B, SCLC NCI-H446, and NCI-H69 cell lines, which were acquired from ATCC (Wuhan, China). The BEAS-2B cell line was cultivated in Dulbecco’s modified Eagle’s medium (DMEM) high-glucose medium (Invitrogen) containing 10% fetal bovine serum (FBS; Invitrogen), and the NCI-H446 and NCI-H69 cell lines were cultivated in RPMI1640 medium (Invitrogen) with 10% FBS. All the cell lines were maintained in an incubator at 37°C and 5% CO2. The total RNA of BEAS-2B, NCI-H446, and NCI-H69 cells was extracted with the TRIzol reagent (Invitrogen). The RNA from these cell lines was transcribed into cDNA by the use of the PrimeScript™RT Reagent Kit with gDNA Eraser (Takara, Japan). The thermocycling protocol involved initial denaturation at 95°C for 30 seconds, then 40 cycles at 95°C for 5 seconds, and 60°C for 30 seconds. Primer sequences applied were as follows:
GAPDH (forward) - 5’-AGAAGGCTGGGGCTCATTTG-3’ and GAPDH (reverse) - 5’-AGGGGCCATCCACAGTCTTC-3’; ZWINT (forward) - 5’-GGAGGAAGCCCAGAGGAAAC-3’ and ZWINT (reverse) - 5’-CTGTCTTACGCTCCCTCACC-3’; NRCAM (forward) - 5’-GAGCGAAGGGAAAGCTGAGA-3’ and NRCAM (reverse) - 5’-ACAATGGTGATCTGGATGGGC-3’. The primers were synthesized by Shanghai Dingguo Biotechnology.
Immune infiltration analysis
The levels of immunocyte infiltration in SCLC tissues with normal lung tissues among the training group were carried out by the use of the cell type identification by estimating relative subsets of RNA transcripts (CIBERSORT) package in R, which determines the infiltration of 22 immune cell types for each sample in the training group. The OmicStudio tool was used to generate a correlation heatmap of different immune cell infiltrations. A level of p<0.05 was established to determine statistical significance.
Further analysis of the correlation in diagnostic genes with different infiltrating immune cells among the training group was carried out using the reshape2, ggpubr, and ggextra packages in R, employing Spearman’s rank correlation.
Results
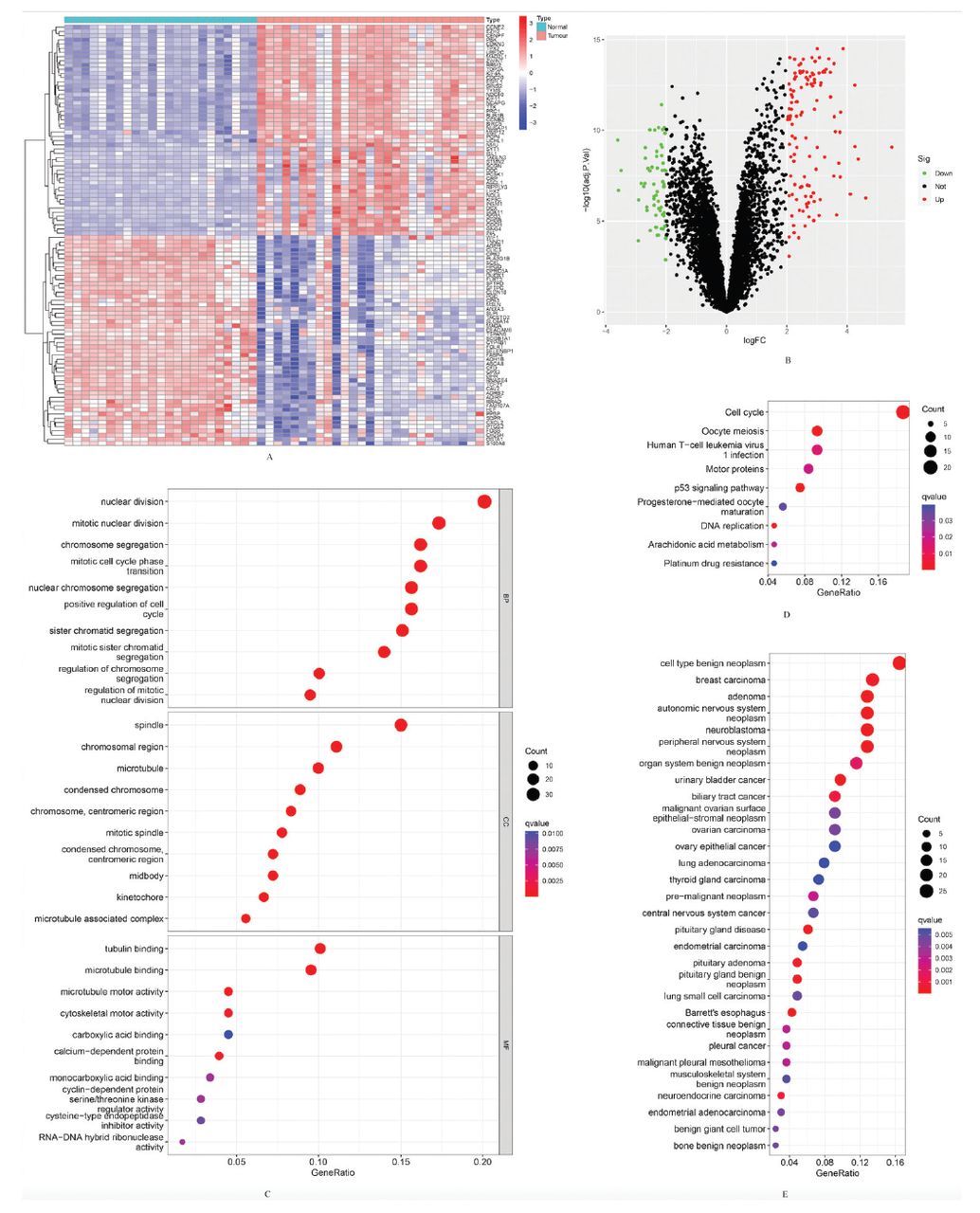
In total, there were 181 DEGs identified, with 119 genes showing upregulation and 62 genes showing downregulation. The results are presented, including a clustering heatmap displaying the top 100 genes (Figures 1A&B).

- Differentially expressed genes (DEGs) between the 2 groups of samples and functional enrichment analyses of them. A) Clustering heatmap of the top 100 DEGs in the training group (red represents relative upregulation, and blue represents relative downregulation of gene expression). B) Volcano plot of DEGs in SCLC tissues with normal lung tissues in the training group (red dots for upregulated genes and green dots for downregulated genes with an adjusted p<0.05 and |log fold change| >2). C) Gene ontology enrichment analysis of functions in the training group. D) Kyoto encyclopedia of genes and genomes enrichment analysis of pathways in the training group. E) Disease ontology enrichment analysis of the training group.
The analysis of GO encompassed 3 components: biological processes (BP), cellular components (CC), and molecular functions (MF). There were 388 BP, 48 CC, and 19 MF enriched in the GO analysis, and the top 10 items were shown (Figure 1C). The DEGs were primarily enriched in BP related to immunity. These processes include leukocyte chemotaxis, myeloid leukocyte migration, and an antimicrobial humoral response. They are also involved in the antimicrobial humoral immune response mediated by antimicrobial peptides, cell chemotaxis, and granulocyte chemotaxis. Additional processes encompass the humoral immune response, defense response to bacteria, and neutrophil chemotaxis. Myeloid leukocyte-mediated immunity, neutrophil migration, and defense responses to fungi were also included. Further processes involve myeloid cell activation in the immune response, antibacterial humoral response, mast cell activation, and myeloid leukocyte activation. There were 9 signaling pathways enriched in KEGG analysis of DEGs (Figure 1D). The DO analysis indicated that DEGs were related to 116 diseases (Figure 1E).
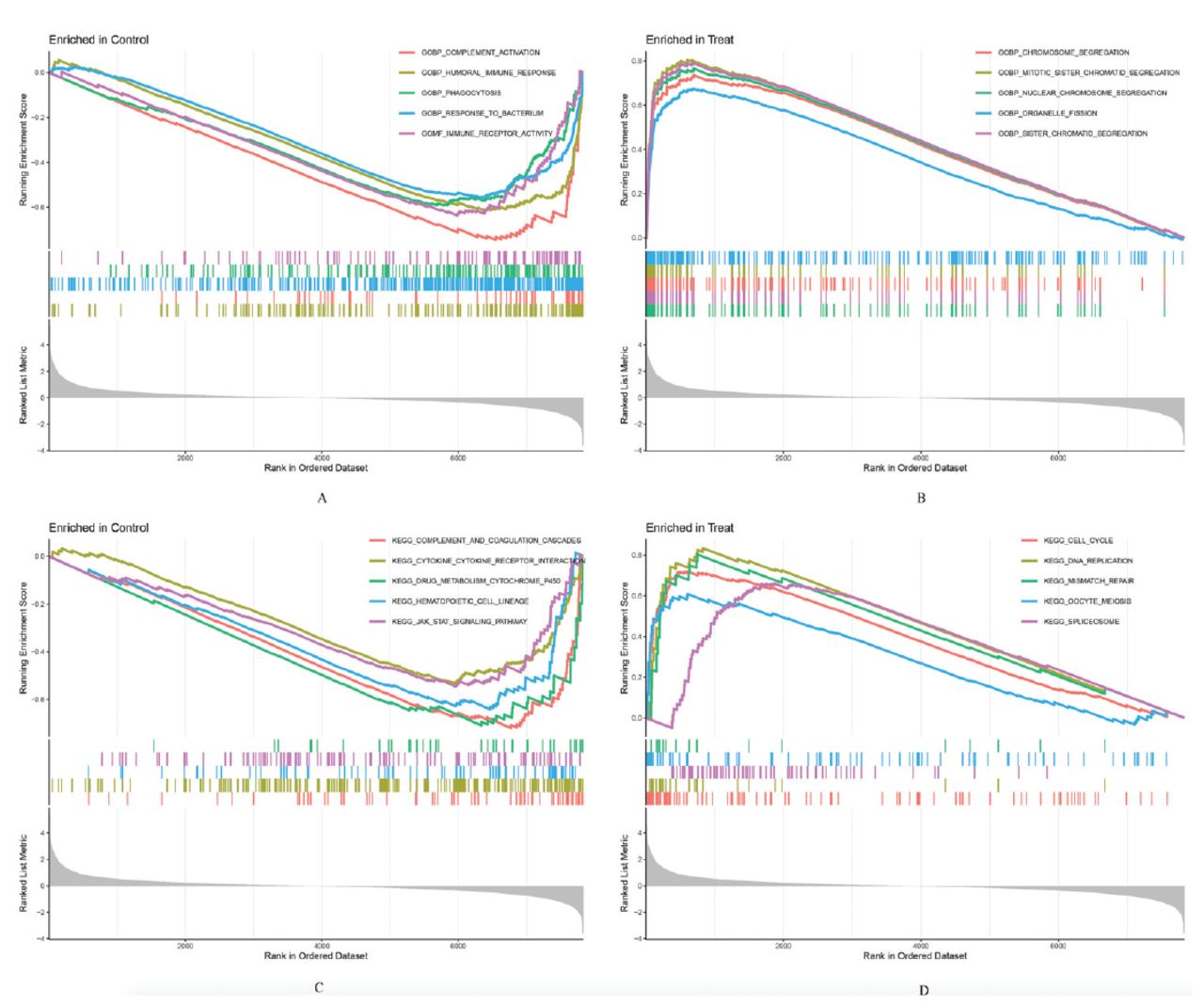
The functional outcomes of GSEA between SCLC tissues and normal lung tissues among the training group showed that multiple items were related to immunity (Figures 2A&B). The GSEA results showed that several pathways were also closely associated with immunity, including complement and coagulation cascades, graft versus host disease, and allograft rejection, etc. (Figures 2C&D).

- Gene set enrichment analysis (GSEA) of functions and pathways in the training group. The top 5 functions of GSEA in: A) normal lung tissues and B) small cell lung cancer (SCLC) tissues, and the top 5 pathways of GSEA in: C) normal lung tissues and D) SCLC tissues.
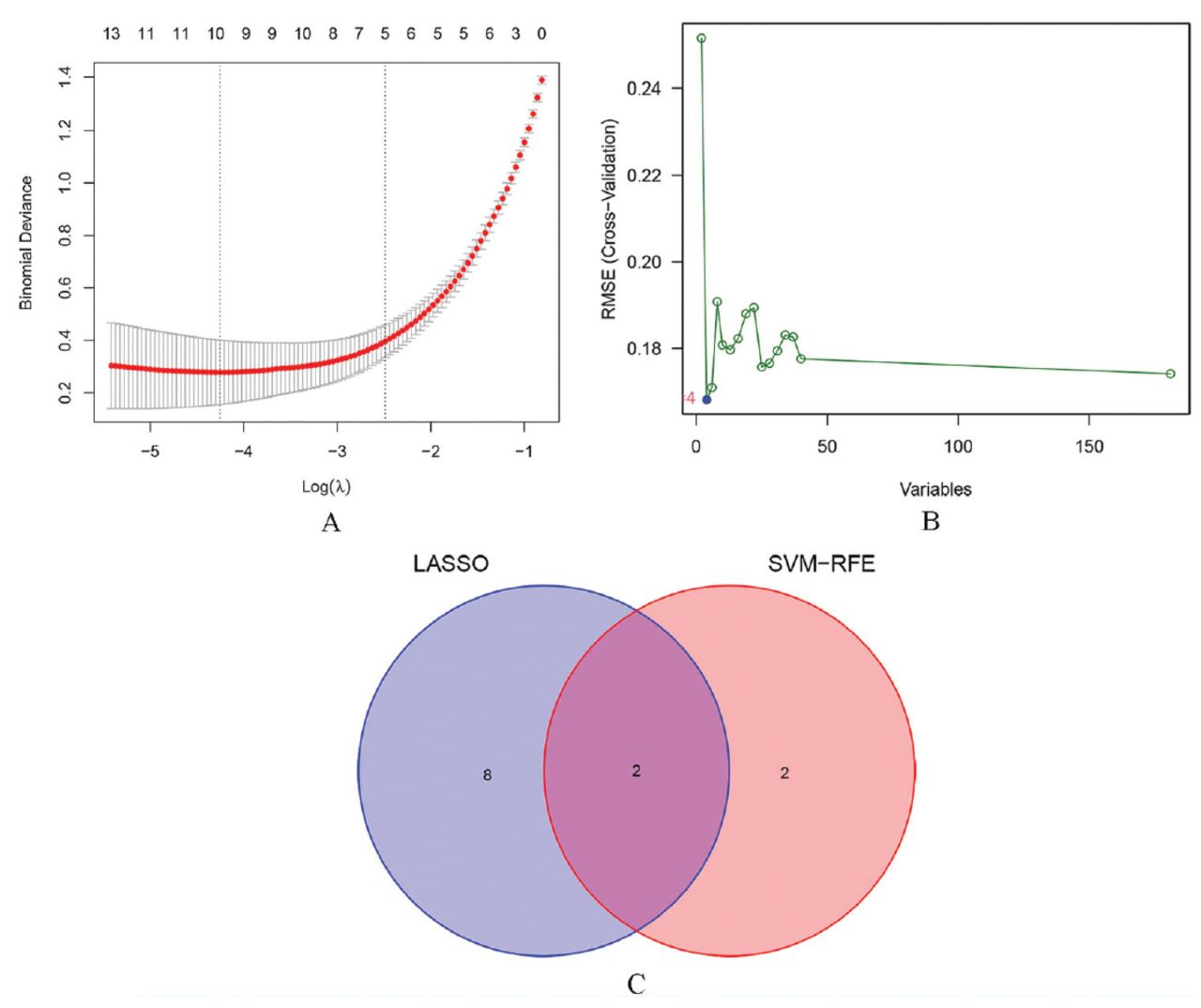
A total of 10 diagnosis-associated genes were identified using the LASSO model: ZWINT, TYMS, PCP4, NRCAM, SOX4, PLA2G1B, CST6, SCGN, PPBP, and CXCL13 (Figure 3A). Four diagnosis-associated genes, RFC4, NRCAM, EZH2, and ZWINT, were identified from DEGs by the use of the SVM-RFE method (Figure 3B). The intersecting section were ZWINT and NRCAM as candidate diagnostic genes (Figure 3C).

- Identification of candidate diagnostic genes. A) Least absolute shrinkage and selection operator regression plot (the X-axis is logλ, and the Y-axis is the cross-validation error). B) Support vector machine-recursive feature elimination algorithm (the X-axis represents a change in the number of genes, and the Y-axis represents a cross-validation error). C) Venn diagram (intersection of genes using 2 machine learning methods). LASSO: least absolute shrinkage and selection operator, SVM-RFE: support vector machine-recursive feature elimination, RMSE: root mean squared error
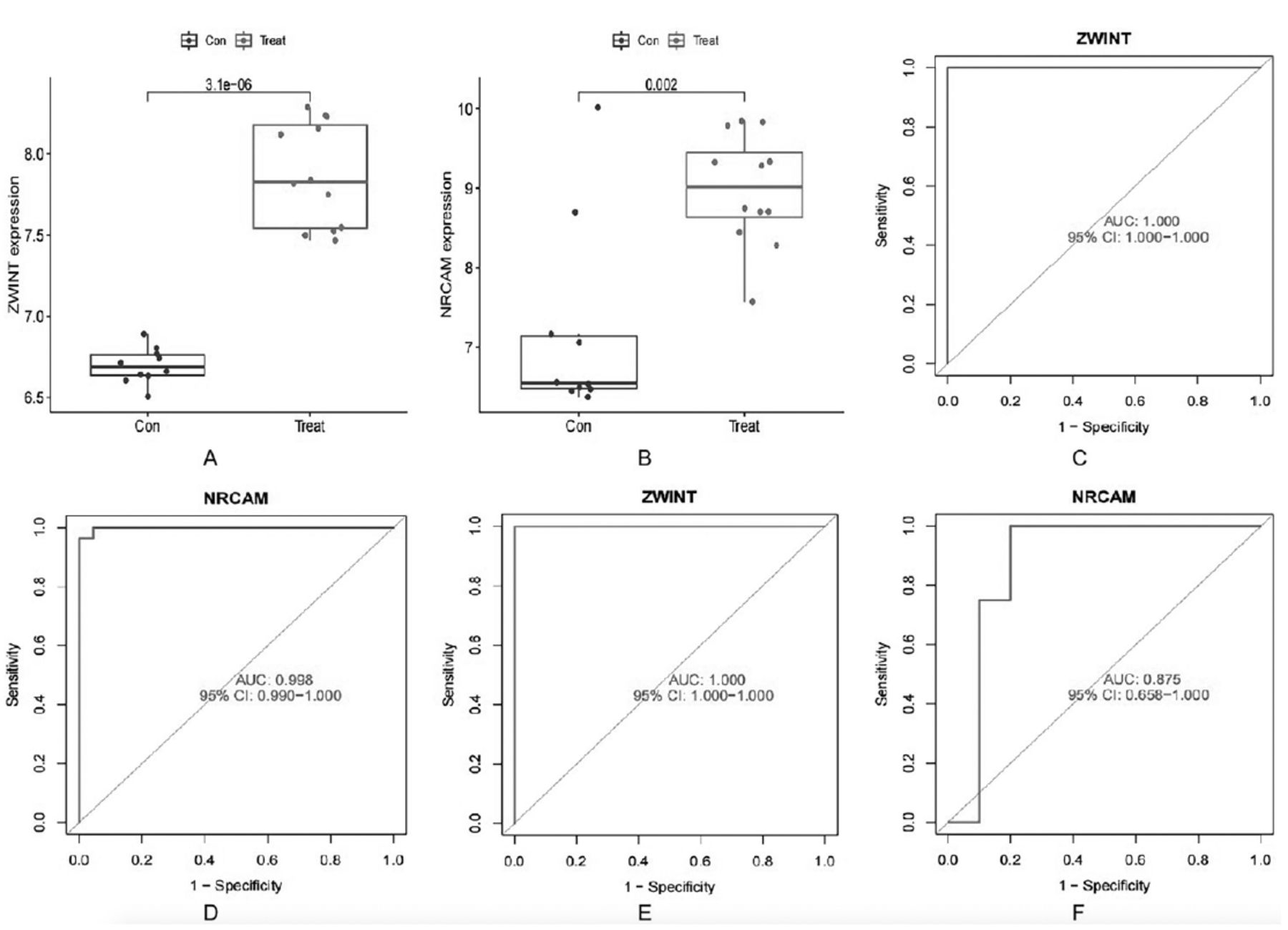
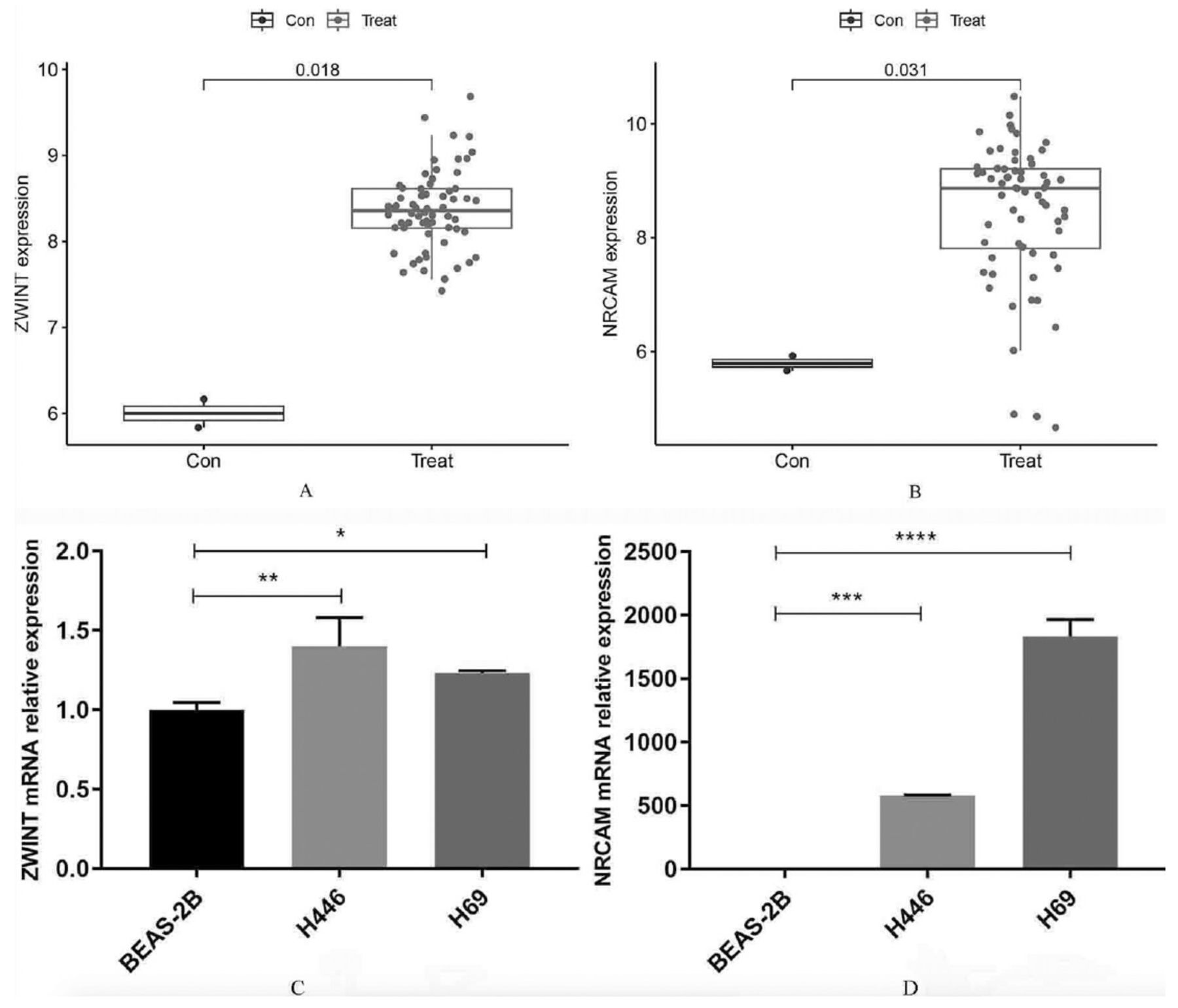
In validation group A, the levels of ZWINT and NRCAM expression were found to be significantly elevated in SCLC tissues compared to normal lung tissues (Figures 4A&B). The area under the ROC curve (AUC) value for ZWINT in the training group was determined to be 1.000 and the AUC value for NRCAM in the training group was determined to be 0.998 (Figures 4C&D). The AUC values were 1.000 and 0.875 in validation group A (Figures 4E&F). The results indicated that all values were greater than 0.80, demonstrating a high predictive accuracy and diagnostic efficacy. Additionally, compared to normal lung cell lines in validation group B, the levels of ZWINT and NRCAM expression were higher in SCLC cell lines (Figures 5A&B).

- Evaluation of the candidate diagnostic genes. A&B) Box plots revealing the expression of ZWINT and NRCAM between small cell lung cancer tissues (treat) and normal lung tissues (con) in the validation group A (p<0.05 represents a significant difference). C&D) The receiver operating characteristic (ROC) curves of ZWINT and NRCAM in the training group. E&F) The ROC curves of ZWINT and NRCAM in the validation group A. AUC: area under the curve, CI: confidence interval

- Further validation of the candidate diagnostic genes and relative expression of them. A) The differential expression of ZWINT and B) NRCAM between small cell lung cancer cell lines (treat) and normal lung cell lines (con) in the validation group B. C) Relative expression of ZWINT and D) NRCAM by quantitative real-time polymerase chain reaction in different cell lines. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001
Using the BEAS-2B cell line as a control, the relative expression of ZWINT and NRCAM in the 2 SCLC cell lines (NCI-H446 and NCI-H69) was analyzed. Compared to the BEAS-2B cell line, the qRT-PCR outcomes revealed that ZWINT and NRCAM were upregulated among these 2 SCLC cell lines. These differences have statistical significance (p<0.05). The detailed results are presented (Figures 5C&D). Based on these results, ZWINT and NRCAM were identified as diagnostic genes.
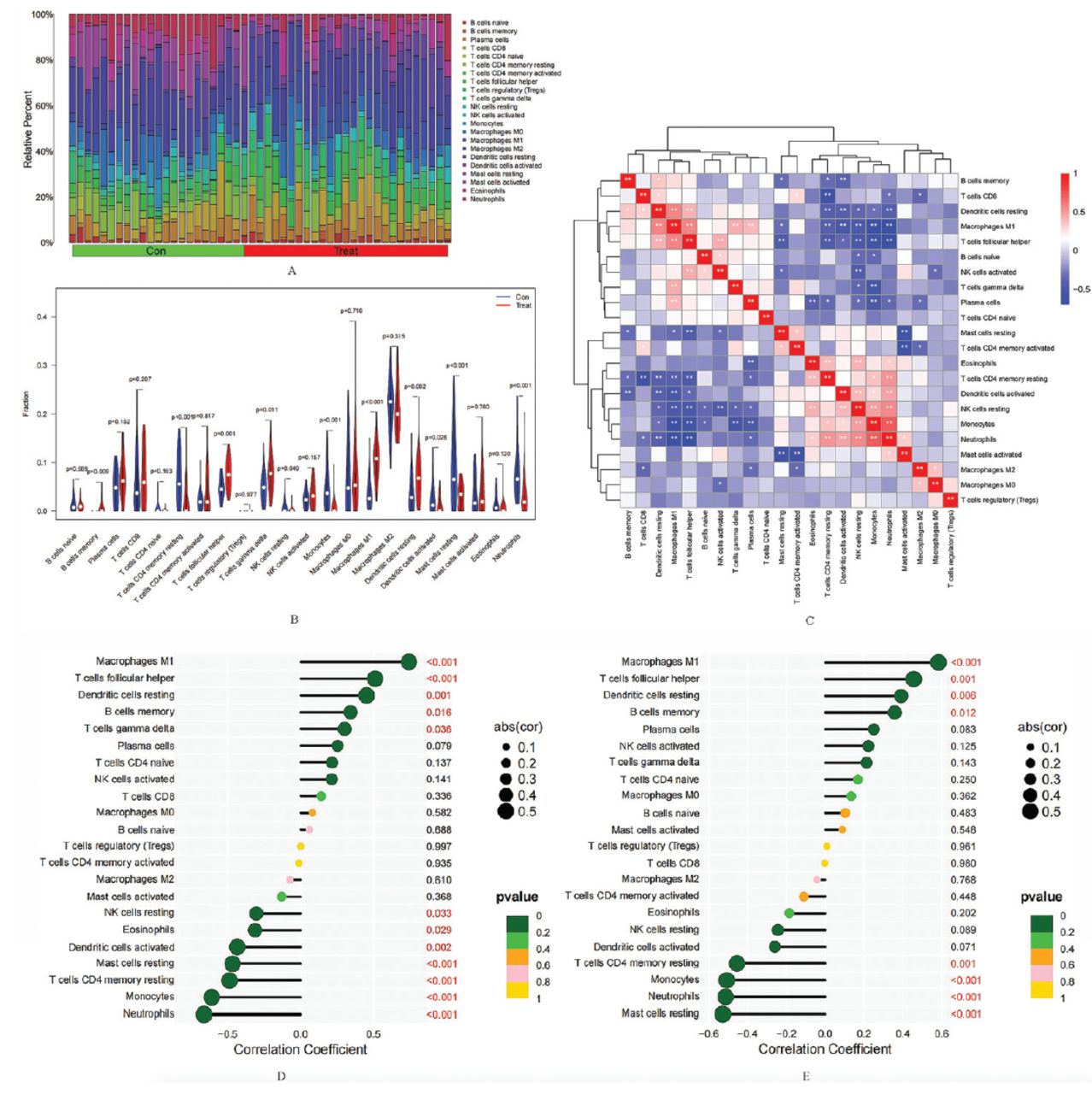
The percentage of 22 different immunocyte infiltrations of each sample was diverse in the training group (Figure 6A). Further analysis revealed that the levels of 11 immune cell infiltrations in SCLC tissues and normal lung tissues were statistically different among the training group (Figure 6B). Compared to normal lung tissues, the SCLC tissues showed elevated levels of M1 macrophages, and resting dendritic cells, etc, whereas lower levels of monocytes, activated dendritic cells, and neutrophils, etc. Correlation analysis between different immunocytes (Figure 6C) indicated that neutrophils were positively related to monocytes (r=0.70), eosinophils (r=0.32), and activated mast cells (r=0.31), and more, while negatively related to follicular helper T-cells (r= -0.67), M1 macrophages (r= -0.66), and plasma cells (r= -0.33), and more. The M1 macrophages were positively related to resting dendritic cells (r=0.57), and plasma cells (r=0.39), and more, and negatively correlated with monocytes (r= -0.68), and resting mast cells (r= -0.31), and more. The above results all have statistical differences. These findings cumulatively indicate that the immunocyte infiltration features of SCLC and normal lung tissue are different and reveal intricate associations among various immune cell infiltrations within the TME.

- Analysis of immunocyte infiltration. A) The relative percentage of different immunocytes infiltration in each sample. B) Violin plot revealing the differences in each infiltrating immunocyte type between small cell lung cancer (SCLC) tissues and normal lung tissues. C) Correlation analysis between different immune cell infiltration levels. Correlation analysis of D) ZWINT and E) NRCAM with different immune infiltrating cells (the X-axis represents the correlation coefficient, and the Y-axis represents the immunocyte names). Con represents normal lung tissue, and treat represents SCLC tissue. A p-value of <0.05 indicates a significant difference.
Correlation analysis revealed that the level of ZWINT expression was positively related to macrophages M1 (r=0.74), and memory B-cells (r=0.34), and more. Conversely, it exhibited a negative correlation with neutrophils (r= -0.66), monocytes (r= -0.61), and eosinophils (r= -0.31), and more. The detailed outcomes are shown in Figure 6D. Furthermore, NRCAM expression levels were a positive association with the infiltration level of macrophages M1 (r=0.58), and memory B-cells (r=0.36), and more. Conversely, it exhibited a negative correlation with neutrophils (r= -0.51), and monocytes (r= -0.51), and more. The above results all have statistical differences. The detailed outcomes were displayed in Figure 6E. These findings indicated a close association in diagnostic genes with immune infiltrating cells.
Discussion
Small cell lung cancer is considered the most malignant type of lung cancer, exhibiting a high rate of cell proliferation, rapid tumor growth, and early metastasis. Despite significant advancements in the number and efficacy of targeted therapies, there have been minimal changes in treatment plans and overall survival for SCLC, which continues to have a poor prognosis.20,21 Recently, immunotherapy has garnered significant attention in cancer treatment.22-24 More and more researches are focusing on novel treatment strategies for SCLC, and progress has been carried out in uncovering its biological properties and microenvironment.25 The infiltration features of immunocyte in the TME are closely associated with the therapeutic effects.26-28 Despite the promising clinical benefits of immunotherapy in treating SCLC, numerous issues remain, and further research is required to clarify them.6 Therefore, in this study, DEGs were identified, and functional enrichment analysis was carried out using bioinformatics tools. The results revealed associations between both tumor and immune responses. The 2 candidate diagnostic genes (ZWINT and NRCAM) for SCLC were identified using LASSO and SVM-RFE methods. Then, the elevated expression levels of ZWINT and NRCAM were validated, and their high diagnostic efficacy was evaluated in both the training and validation groups. Moreover, their relatively high expression levels were carried out by qRT-PCR. Immune cell infiltration and correlation analyses indicated notable variances in the features of infiltrating immune cells, as well as strong connections in diagnostic genes with immune cell infiltration.
This study identified 181 DEGs, with 119 genes showing upregulation and 62 genes showing downregulation. The GO analysis indicated that DEGs enriched in BP were related to immunity. The results of DO analysis included cell type benign neoplasms, breast carcinomas, adenomas, autonomic nervous system neoplasms, neuroblastomas, and SCLC. The GSEA in function and pathway between SCLC tissues and normal lung tissues in the training group displayed that multiple items were related to immunity. These findings suggest that DEGs are associated with tumors and immunity.
The ML methods are commonly used in clinical decision-making.29 The LASSO regression and SVM-REF models are 2 common models in ML. Transcriptome sequencing data are usually high-dimensional with many variables (gene expression levels) and samples (different cell types or disease states), and traditional linear regression methods cannot process these data efficiently. The LASSO regression is a new linear regression method that selects genes associated with a physiological phenomenon or a disease by minimizing the sum of absolute values, which can effectively handle high-dimensional data and select the most important genes for functional prediction.30 The LASSO is a commonly used method, and its clinical efficacy has been confirmed.31,32 The SVM-RFE is an ML method based on support vector machines, which can be utilized in bioinformatics to extract feature genes from the expression matrix of differential genes. Based on their setup of grouping variables, they can ultimately achieve the goal of identifying optimal variables through the feature vectors generated by the SVM. This ML method was applied for screening characteristic genes.33 The SVM-RFE model is also widely used to screen diagnostic markers for conditions such as tumors, cardiovascular diseases, and immune disorders.33-35 To identify potential diagnostic genes for SCLC, LASSO, and SVM-RFE algorithms were constructed, with 10 genes identified by the former and 4 genes by the latter. The intersection region (ZWINT, NRCAM) was regarded as a candidate diagnostic gene. In validation group A, the expression levels of ZWINT and NRCAM were found to be significantly elevated in SCLC tissues compared to normal lung tissues. In the training group and validation group A, the AUC values suggested that they exhibited a higher predictive effect and diagnostic efficacy. In the validation group B, the SCLC cell lines exhibited elevated levels of ZWINT and NRCAM expression compared to normal lung cell lines. Additionally, compared to the BEAS-2B cell line, qRT-PCR outcomes showed that the levels of ZWINT and NRCAM expression were upregulated in these 2 SCLC cell lines (p<0.05). Therefore, ZWINT and NRCAM were considered diagnostic genes.
Immune infiltration analysis was employed to characterize the composition of immune cells within the human microenvironment and to identify which specific immune cells play a crucial role in disease development. The CIBERSORT is widely used for this purpose because, among the various immune cell infiltration databases, it utilizes linear support vector regression for deconvolution analysis. This user-friendly method provides a comprehensive range of immune cell classes and covers 22 types.36 In this study, an analysis comparing immunocyte infiltration in SCLC with normal lung tissues revealed diverse proportions of various immune cells in each case. Additionally, notable variances in the infiltration levels of 11 immunocyte types between SCLC and normal lung tissues were observed in the training group. Immune cells are essential constituents of TME and have important roles in tumorigenesis, which may have tumor-antagonizing or tumor-promoting effects.37,38 The TME is a complex and diverse system, and the formation, progression, and metastasis of cancer are closely linked to the internal and external conditions surrounding the cancer cells.8 The heterogeneous malignant components of the TME may be linked to angiogenesis, nutrition/blood supply, and tumor metastasis, highlighting the recurring characteristic of tumor cell heterogeneity in SCLC. Consequently, the heterogeneity of malignant cells reflects variations in the interactions among TME components, SCLC subtypes, and varied responses to drugs.13 The high ITH and intricate nature of cancer cells contribute to drug resistance, thereby posing significant challenges in cancer therapy.39 Correlation analysis between different immunocyte infiltrations indicated complicated interrelationships in the TME, and the outcomes were in consistent with the research of Zhong et al40 in lung cancer and normal tissues. These findings may provide new insights for immunotherapy of cancer.40 Correlation analysis of the diagnostic biomarkers with immunocyte infiltration indicated a close and comprehensive relationship between them, suggesting mutual interactions that impact the immune infiltration features of the TME. These findings are consistent with those of Xie et al41 in gastric cancer and normal tissues. The above results demonstrated notable disparities of immune cell infiltration characteristics in SCLC with normal lung tissues, revealing complicated correlations between immune cells infiltration of TME. These differences could be associated with the prognosis and immunotherapy outcomes.
The ZWINT was a crucial component of the centromere complex necessary for the mitotic spindle checkpoint, which is associated with centromere function, and it is significantly upregulated in breast cancer tissues, indicating a poor prognosis for patients.42 The ZWINT exhibits high expression in lung adenocarcinoma tissues and is associated with unfavorable prognosis in lung adenocarcinoma patients. The knockdown of ZWINT inhibits proliferation, migration, invasion, and colony formation in NCI H226 and A549 cells, which could become a new target for lung cancer therapy.43 Therefore, the high expression of ZWINT in SCLC could be associated with poor prognosis and therapy. The NRCAM is a member of the immunoglobulin superfamily, its expression is related to low-grade neuroblastoma in children and could play a part in the early development of neuroblastoma.44 NRCAM is highly expressed in papillary thyroid cancer and may be a possible diagnostic marker and therapeutic target for this disease.45 Consequently, the elevated expression of NRCAM in SCLC may act as a diagnostic marker and have therapeutic implications.
ZWINT and NRCAM have significant potential in the diagnosis of SCLC. Therefore, the expression of these 2 proteins in the serum can be detected by ELISA. The expression levels of serum neuron specific enolase and progastrin-releasing peptide, which are currently common tumor markers for the diagnosis of SCLC, can be jointly detected, thus improving the diagnostic efficiency of SCLC, including early diagnosis, and decreasing the misdiagnosis rate. Furthermore, these 2 proteins are highly expressed in SCLC tissues. They can be validated through a series of experiments, including cell, animal, and clinical studies, to identify potential molecular targets for SCLC treatment.
Study limitations
First, owing to the lack of prognostic information in the GEO database, a prognostic analysis of the 2 diagnostic genes could not be carried out. In future research, paraffin-embedded tissues from over 100 patients with SCLC will be collected at the hospital for immunohistochemical staining of these 2 proteins. These patients will be followed up to determine whether high expression levels of these proteins are associated with prognosis. Second, further analysis of the 2 diagnostic genes related to immune cell infiltration in SCLC subtypes was not carried out. Lastly, clinical samples from SCLC patients and controls were not collected for in-depth validation. Therefore, serum specimens from patients with SCLC, NSCLC, and benign lung nodules will be collected for ELISA to measure the expression of these 2 proteins. Diagnostic efficacy testing will be carried out to assess their potential as diagnostic markers. If the diagnostic efficacy is promising, a multicenter collection of serum specimens will be carried out for further validation, laying the groundwork for clinical application.
In conclusion, this study identified 2 diagnostic genes, ZWINT and NRCAM, which are correlated with immune cell infiltration through the integration of bioinformatics analysis and ML algorithms. These genes could serve as potential diagnostic biomarkers and offer possible molecular targets for immunotherapy in SCLC.
Acknowledgment
The authors gratefully acknowledge Home of Researchers for their English language editing.
Footnotes
Disclosure. This study was supported by the Key Science and Technology Research Project of Jiangxi Provincial Education Department, Jiangxi, China (GJJ220C119).
- Received March 4, 2024.
- Accepted July 4, 2024.
- Copyright: © Saudi Medical Journal
This is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.