


Abstract
Renal lymphangiectasia (RL) is a rare condition in which lymphatic vessels are dilated giving rise to cyst formation in peripelvic, perirenal and intrarenal locations. Knowledge about RL is limited and based upon individual case reports. This can be genetic or acquired. There is no significant association with any gender or age. It can be manifested as focal or diffuse forms and can be unilateral or bilateral. Most of the cases present with abdominal or flank pain. The diagnosis is based on radiological imaging. Due to rarity of diseases, it has potential to be misdiagnosed as other cystic disease of kidneys. The treatment is mainly conservative but prolonged follow up for associated complications like hypertension and renal vein thrombosis is required. We have presented a case of bilateral renal lymphangiectasia with the review of available literature.
Renal lymphangiectasia (RL) is a rare condition, constituting only 1% of all lymphangiectasias.1,2 Given its infrequency, most of our understanding regarding its etiopathogenesis and natural history is derived from case reports.3,4 Renal lymphangiectasia may manifest unilaterally or bilaterally, without a specific predilection for age or gender.1-3 The condition likely arises from miscommunication within the renal and retroperitoneal lymphatics, leading to the dilatation of lymphatics and cyst formation in and around the kidneys.1,2,5 Failure to recognize this entity leads to misdiagnosing it as more common cystic lesions, which necessitate entirely different management.4,6,7 In this context, we present a case of bilateral renal lymphangiectasia, adding to the limited literature already available as only 33 such cases have been reported in the adult population.8
Case Report
A 55-year-old Saudi female presented to the outpatient department with a 10-year history of flank pain. She was non-hypertensive and non-diabetic, with no significant medical or family history.
Clinical findings
She complained of bilateral flank pain. The pain was mild, colicky, and non-radiating, associated with mild dysuria without hematuria, fever, weight loss, or abdominal distension. On examination, she was of average built and well-oriented, with stable vital signs within normal ranges for her age. Abdominal examination revealed mild tenderness in both lumbar regions, with no rebound tenderness, guarding, or visceromegaly observed.
Diagnostic assessment
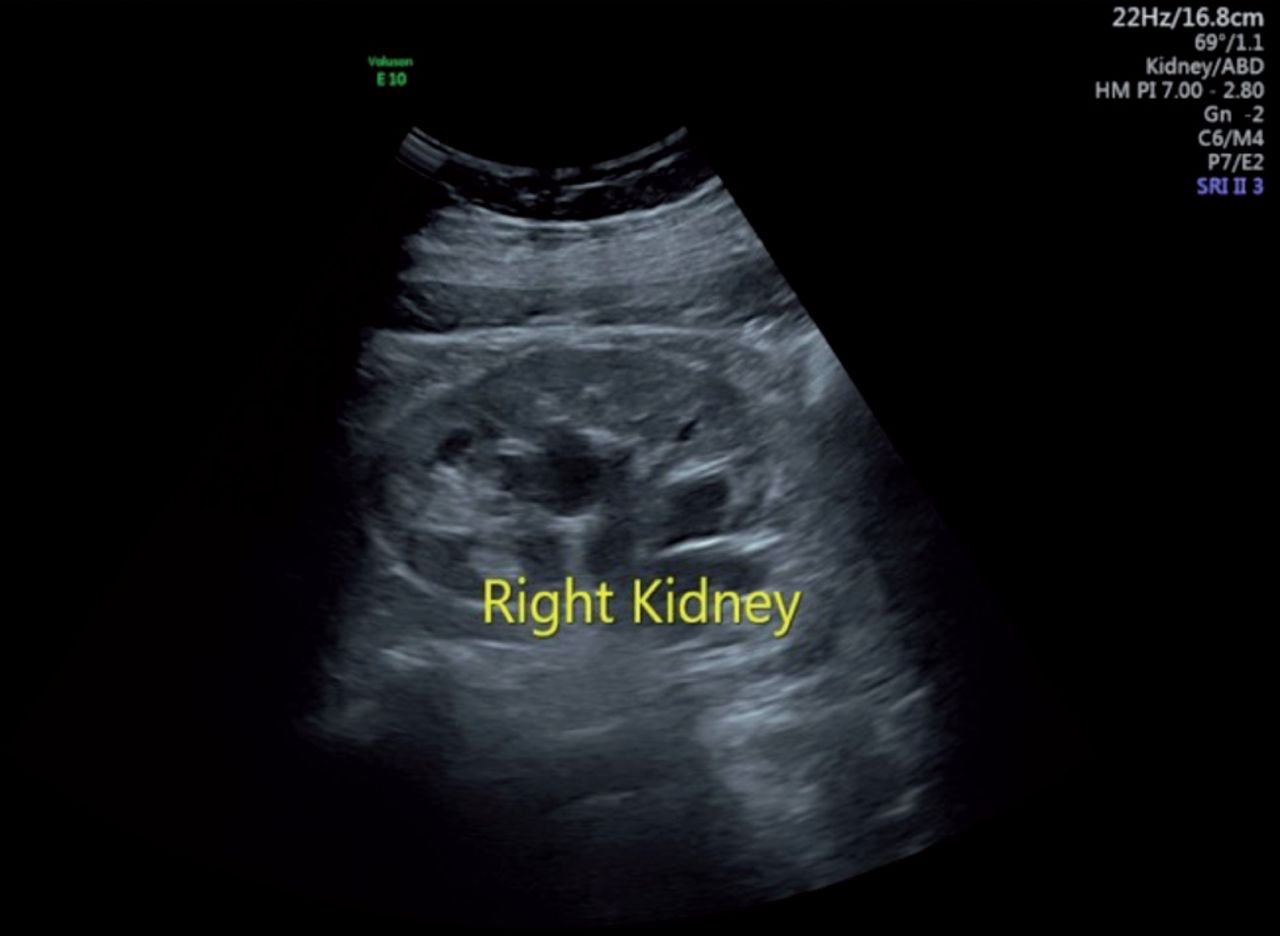
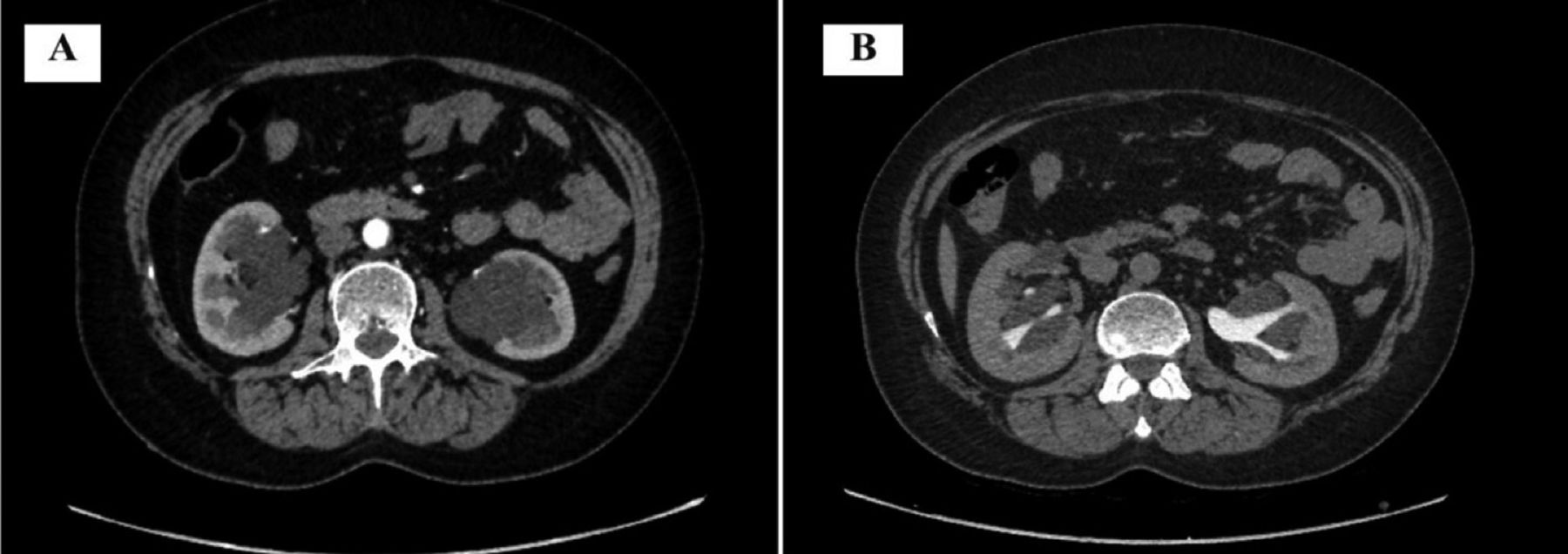
Routine investigations showed a hemoglobin level of 11.6 g/dL and a random blood sugar (BSR) of 6.78 mmol/L. Renal function tests (RFTs), liver function tests (LFTs), complete urine examinations, and other tests were unremarkable. Ultrasonography of the kidney, ureter, and bladder (KUB) revealed normal size, outline, and parenchymal echotexture of both kidneys. Bilateral pelvicalyceal systems showed dilatation (Figure 1), with no stones or masses identified in the ureter or bladder. Confirmatory diagnosis and ruling out sinister pathology were pursued through contrast-enhanced computed tomography (CT). Computed tomography findings showed normal shape, size, and position of both kidneys, with intact corticomedullary differentiation. Hypodense filling defects in the pelvis with a density ranging from -3 to +7.25 HU (Hounsfield unit) consistent with lymphatic tissue was observed (Figure 2A). Drainage phase showed that pelvicalyceal system was compressed, and mild dilatation of the proximal pelviureteric junction was noted (Figure 2B). The middle and distal thirds of the ureter were normal, and the urinary bladder exhibited normal blood flow, with no stones or space-occupying lesions. Early phase showing hypodense filling defect in the pelvis with bilateral squashed pelvicalyceal system in delayed phase is characteristic finding of RL thus a definitive diagnosis of RL was established.

- Ultrasonography showing dilatation of pelvicalyceal system in right kidney.

- Axial section of computed tomography with contrast image. (A) Early phase showing hypodense filling defect in the pelvis of both kidneys. (B) Drainage phase showing bilateral squashed pelvicalyceal system by lymphatic mass.
Therapeutic intervention
The patient was managed conservatively and advised periodic follow-up.
Follow-up and outcomes
Patient is doing well without any associated complications or comorbidities.
Timeline of the patient first clinical presentation to follow up is shown in Figure 3.

- Timeline summarizing the patient’s profile, history, examination, routine investigations, ultrasonography, computed tomography with contrast, diagnosis, treatment, and follow up.
Discussion
Our patient was diagnosed with bilateral RL based on characteristic radiological findings observed in a contrast-enhanced CT scan. This rare, benign, developmental malformation of renal lymphatics has been documented in the literature under various names, such as RL, renal lymphangiomatosis, renal lymphangioma, renal lymphatic malformation (RLM), peri-pelvic lymphangiectasia, renal sinus polycystic disease, and renal hygroma.3 Renal lymphangiectasia is the preferred term, as it accurately reflects the nature of the disease as a malformation rather than a tumor.1,5
Lymphangiectasia is usually found in children, most commonly involving the head and neck regions (approximately 70%), followed by the chest and axillary region (20–25%).4,5 Only about 5% of cases are located in internal organs, with kidneys accounting for a rare 1%.1,2,4,5 The rarity of the disease limits our understanding of its etiopathogenesis, incidence, prevalence, and natural history.4,6,8 Clinical presentation and imaging findings are primarily derived from isolated case reports or case series.3-6
The cystically dilated lymphatics of RL can be in the perirenal, peripelvic, and intrarenal regions.5,6 Radiologists, playing a crucial role in diagnosis, must be vigilant, as RL may be misdiagnosed as more common cystic lesions, such as hydronephrosis, polycystic kidney disease, and parapelvic cysts.1,3,5 Proper diagnosis is essential for guiding therapeutic decisions.4,6,7
The data indicates that RL has an equal incidence in males and females.1-3,6 Renal lymphangiectasia can manifest in both pediatric and adult populations,with children more frequently presenting with ascites, signifying a more severe form of the disease.1-3,6,8,9 Renal lymphangiectasia can be focal or diffuse and may be located in the perirenal, peripelvic or intrarenal region in one or both kidneys.3,5,6,9 By 2022 a total of 104 cases of RL had been reported globally, 5 cases were from Saudi Arabia.8 Our cases of RL is bilateral and in an adult, only 33 such cases had been reported earlier.8
The etiology and pathophysiology of RL remain elusive, mechanisms including genetic, developmental, or acquired factors were proposed in various case reports.2,3,5,7-9 A prevailing hypothesis suggests developmental malformations and miscommunication between renal and retroperitoneal lymphatics, resulting in cystic dilatation of lymphatic vessels.1-4,7,9,10 Some studies noted familial clustering of RL cases, while others proposed inflammation, scarring of lymphatic vessels with obstruction, and dilatation as potential mechanisms.5,8
Most RLs are asymptomatic; up to 16% of cases are discovered incidentally through radiological imaging.8 The most common presenting symptoms include pain in the abdomen or flanks, hematuria, and hypertension.5,8 Other symptoms may involve proteinuria, fatigue, weight loss, and fever.1-3,5,8,9 More severe cases can manifest with or be complicated by the rupture of cysts, intracystic hemorrhage, superimposed infection, renal vein thrombosis, ascites, hypertension, or renal failure.1-3,5,8,9 Polycythemia, hemangioma, and glomerulonephritis have also been reported to coexist with RL.2,5,8,9 Our case presented with flank pain without any associated complications.
Clinicoradiological correlation rules out other differential diagnoses and confirms the diagnosis of RL.2,4,5,7 Ultrasonography (USG) and CT with or without contrast enhancement are the most widely used radiological investigations for this purpose.5 While earlier, biopsy and surgical pathology were being used for diagnosis, this invasive approach is now discouraged.5 Few suspicious cases can be confirmed by needle aspiration and cytological examination of chylous fluid.3,4,9
Typically, USG reveals anechoic lesions with increased transmission, and thin-walled cysts, with or without septations in the renal sinus or perirenal location.1,4,7 Computed tomography has emerged as the preferred modality for diagnosing RL. Uncomplicated cases exhibit characteristic findings of well-defined, multiseptated, low-attenuation (0-20HU) lesions in the peripelvic, perinephric, or intrarenal location.1,4,10 Triple-phase contrast-enhanced CT scans demonstrate no opacification of cysts during the delayed phase, a characteristic finding confirming the diagnosis of RL and distinguishing it from hydronephrosis.2,3,7,10 On magnetic resonance imaging (MRI), cystic lesions appear hypointense on T1-weighted images and hyperintense on T2-weighted images.1,5,7 T2-weighted images also reveal increased cortical and decreased medullary intensity.5,7 Lymphoscintigraphy, a recent introduction, aids in detecting abnormal lymphatic flow.1 In our case, USG raised suspicion of bilateral cystic lesions in the pelvicalyceal region, which was subsequently confirmed by a CT scan.
Cytological examination of aspirated fluid is reserved for suspicious cases.1,3 The gross appearance may not be milky or chylous, as it does not involve mesenteric drainage.4 Lymph from renal origin characteristically exhibits only a few cells, small amounts of protein, and fat globules with elevated renin levels.4,7
Several differential diagnoses are considered depending on the age, clinical features, and appearance of the disease. The most common mimickers include hydronephrosis, polycystic disease, multilocular cystic nephroma, lymphoma, and nephromblastomatosis.1-3,9
Due to rarity, the natural history of the disease is unclear.5 Renal lymphangiectasia may have rapid progression, regression, or spontaneous remission.5,10
The choice of treatment depends on presenting symptoms; however, existing literature does not conclusively recommend one treatment modality over another.8 Most asymptomatic cases do not require treatment, and patients with mild symptoms, such as in our case, are managed conservatively.2,9,10 Analgesics are prescribed for pain, antihypertensives for arterial hypertension, or diuretics to control ascites.1,8 For large or rapidly growing lesions causing pressure symptoms, percutaneous aspiration with or without sclerotherapy or marsupialization can be performed.1,2,4,8,10 Close follow-up is required in all cases to monitor potential complications like hypertension, renal vein thrombosis, or renal failure.3,7,10 For severe, uncontrollable, or complicated cases like renal vein thrombosis or Page kidney, nephrectomy can be considered.1,3,4,8 However, nephrectomy is usually avoided as it may lead to an increase in the size of lymphatic cysts in the contralateral kidney.7,9
In conclusion, RL is a rare disease that requires differentiation from other conditions based on characteristic radiological features. Treatment is conservative, with periodic follow-ups for hypertension and renal insufficiency. Radiologists and physicians should be familiar with this rare entity and vigilant in achieving a proper diagnosis and treatment in order to avoid unnecessary and invasive diagnostic and therapeutic modalities.
Acknowledgment
The authors are thankful to the Deanship of Graduate Studies and Scientific Research at University of Bisha for supporting this work through the Fast-Track Research Support Program. We would like to thank Editage (www.editage.com) for the English language editing.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received December 26, 2023.
- Accepted April 16, 2024.
- Copyright: © Saudi Medical Journal
This is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.