


Abstract
L-2-Hydroxyglutaric aciduria (L-2-HGA) is a rare disorder. The patients have psychomotor retardation, ataxia, macrocephaly, and epilepsy usually in childhood. We present a case of L-2-HGA who developed dystonia in the third decade of life. The family reported symptoms of progressive psychomotor regression since childhood. On assessment, the patient had mild impairment of higher mental functions, mild exotropia, and right-hand dystonia. Brain MRI revealed diffuse bilateral symmetrical subcortical white matter hyperintense signals. 2-hydroxyglutaric acid in urine was elevated and the whole genome sequencing revealed a homogeneous pathogenic variant of the L-2-hydroxyglutarate dehydrogenase (L2HGDH) gene. The prognosis was explained to the caregivers. Patients with mild phenotype L-2-HGA can remain undiagnosed until adulthood. Cases of dystonia even without complaints of epilepsy should be investigated by MRI -brain, urine test and genetic testing to rule out L-2-HGA.
L-2-hydroxyglutarate dehydrogenase (L-2-HGDH) is a mitochondrial membrane-related metabolic enzyme. Mutation in human genes causes a neurometabolic disorder called L-2-hydroxyglutaric (L-2-HG) aciduria (L-2-HGA).1 It is an autosomal recessive type of genetic pattern but mutation causes a wide variety of phenotypes.2 It is usually observed in children, slowly progressive, and causes cerebella ataxia, mild to severe mental retardation, signs of extrapyramidal and pyramidal involvements and seizures. The urine and cerebrospinal fluid show increased levels of L-2-HG.3 Autism spectrum disorder (ASD) is linked to different inborn errors of metabolism including organic acidurias like propionic aciduria and L-2-HG aciduria.4
The neurological affections of L-2-HG aciduria are confirmed by Brain magnetic resonance imaging (MRI). They show nonspecific subcortical white matter loss, centripetal subcortical leukoencephalopathy, cerebellar atrophy, and changes in dentate nuclei.5 The clinical and radiological findings of L-2-HGA are confirmed by targeted L-2-HGH and L-2-HGA sequencing to identify the pathological mutation.6
Although muscle weakness and cardiomyopathy have been associated with L-2-HG aciduria, dystonia is a less recognized feature of this ailment.7 Few cases of L-2-HG aciduria-related dystonia are reported in children and teenagers.8 However, dystonia in the third decade in these patients is not common.
We present a comprehensive workup of a patient of L-2-HGA who presented with dystonia of the unilateral right upper arm in the third decade of life while neurological signs and symptoms existed since early childhood.
Case Report
Patient information
A 27-year-old female presented in June 2023 with abnormal right upper limb movement for 4 months. This abnormal movement progressed slowly, worsened with stress and physical activity, and affected her daily activities. The family members reported that she suffered since early childhood and they noted signs of progressive psychomotor regression therefore she could not attend regular school. She did not have any complaints of regular or chronic headaches, visual disturbances, episodes of loss of consciousness or convulsions, urination problems, or constipation. She is mobile independently and can even climb the stairs. She can eat, drink, use the bathroom, and wash herself without help. She was born after a full-term pregnancy and through a normal spontaneous vaginal delivery with no family history of similar conditions among other siblings (2 males and 5 females). There was no history of consanguinity between her parents.
Clinical findings
On examination in our clinic, we noted that the patient had a normal head circumference, without any dysmorphic features, mild impairment in higher mental functions with an intelligence quotient score of 65. Ophthalmic evaluation suggested a mild grade of exotropia but had a normal posterior segment on direct fundoscopy (Heine, Germany). She had normal extraocular movements in all 8 cardinal directions. She had a scanning speech but no dysarthria. All deep tendon reflexes were brisk, with bilateral extensor plantar response and normal power in all limbs. Mild spasticity was noted in the upper and lower limbs. Intention tremor was noted bilaterally and was more prominent on the right upper limb. Right-hand dystonia mainly occurs in the 4th and 5th fingers and is exaggerated by voluntary movements and sustained postures against gravity. There was no null point or voluntary maneuver that temporarily reduced the severity of dystonia in the right hand. There was no mirror dystonia in the left hand. The finger-nose test on both sides was normal. She had an abnormal heel-chin test with an abnormal tandem gait. A mildly ataxic spastic gait was also noted. The laboratory tests for blood, liver functions, renal functions, and thyroid function tests were normal.
Diagnostic assessment
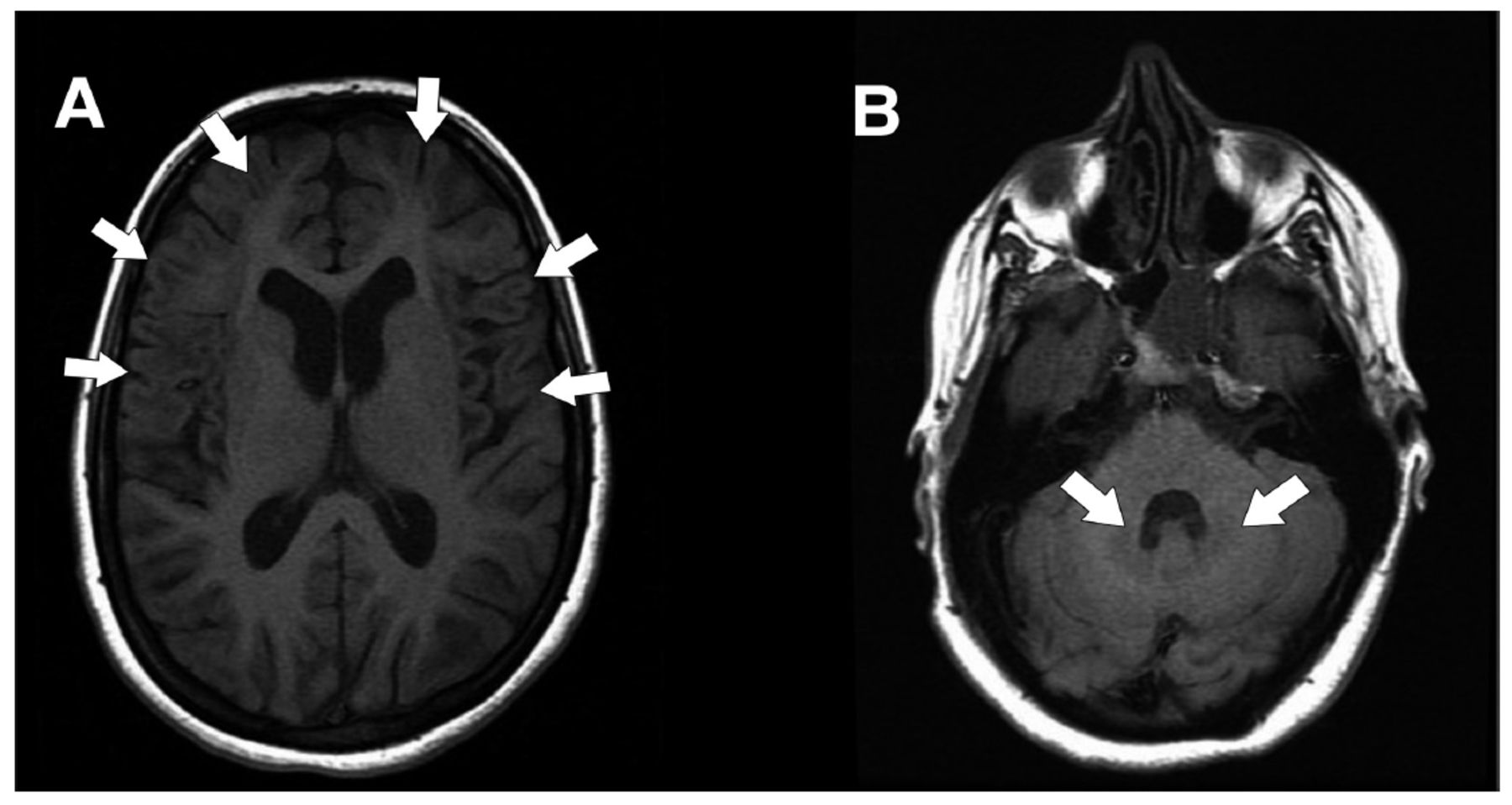
The MRI of the brain (Figure 1) revealed generalized atrophic changes that were predominant in the frontal and parietal lobes. A mild ventricular dilatation and diffuse bilateral symmetrical predominantly subcortical white matter were shown with hyperintense signals in T2 and fluid attenuated inversion recovery (FLAIR) protocols extending to the deep white matter. These involved the bilateral caudate, basal ganglia, and dentate nuclei while sparing the thalamus and brainstem.

- Magnetic resonance axial fluid attenuated inversion recovery (FLAIR) images showing bilateral symmetrical frontal and parietal cortical, subcortical, basal ganglia (A) and dentate nuclei (B) hyperintense signals.
We performed qualitative and quantitative organic acid screening of urine. The urinary excretion of 2-Hydroxyglutaric acid was elevated (769 mmol/mol of creatinine; reference: 0–30 mmol/mol of creatinine). The level of 3-Hydroxyglutaric acid was also elevated (72 mmol/mol of creatinine; reference: 0–8 mmol/mol of creatinine). This confirmed the diagnosis of L-2-HGA. The whole genome sequencing (WGS) showed a homozygous pathogenic null variant in the L-2-HGDHgene, online Mendelian Inheritance in Man registry (609584), Genomic coordinate (Chr14:50267814 G>A), ID Transcript (NM_024884.3), Human Genome Variation Society Nomenclature (c.1003C>T), protein change (p.(Arg335*), location (8/10), zygosity (homozygote), function (stop-gained), high impact and clinically relevant variation database (pathologic) established the genetic diagnosis of autosomal recessive L-2- HGA. Accordingly, the American College of Medical Genetics and Genomics criteria of this condition was (PVS1, PS4, PM2_SUP).
Therapeutic intervention and follow up
The patient and her relative were counseled about the slow progression of the condition and the need for regular follow-up. A therapeutic trial of riboflavin injection was suggested but the patient refused.
Informed consent
Patient and caretaker provided informed written consent in Arabic for using MRI images for the research and publication.
Discussion
This is an unusual case of L-2-HG that presented with right arm dystonia-related symptoms for the first time and when the consultant of the neurology unit investigated, they established hereditary, genetic etiology, and confirmed the diagnosis with the help of brain MRI, urine investigations, and whole genome sequencing.
Dystonia is a common movement disorder. In the present case, it is focal, adult-onset, and secondary to genetic metabolic disorder. The diagnosis mainly clinical is assisted by urine investigation, MRI of the brain, and genetic evaluation.9 Developmental delay, cerebellar ataxia, and epilepsy are the most common clinical features of L-2-HGA. Extrapyramidal features are present in one-third to half of these reported cases. They manifest mainly as tremors but dystonia; generalized, segmental, and task-specific were also reported in patients with L-2-HGA.8
The urinary excretion of 2-hydroxyglutaric acid (769 mmol/mol of creatinine) is significantly lower in our patient than the mean of patients with similar null mutations (1,916 mmol/mol of creatinine) as measured by Steenweg et al.10 A correlation of excretion levels with disease severity however was not demonstrated.
Neuroimaging was a crucial step in identifying the neurometabolic disorder and confirming the diagnosis with biochemical and genetic tests. The mild atrophic changes in MRI are consistent with the mild phenotype shown in this case. The MRI features included the T2W and FLAIR symmetrical hyperintensities in the bilateral basal ganglia, cerebellar dentate nuclei, and bilateral fronto-temporo-parietal subcortical white matter, while the brainstem and thalami were spared. A close correlation between the clinical progression and the extent of changes on MRI has been reported.5
The mutation detected in our patient is a null mutation (DNA variant c.1003C > T, protein variant p.[Arg335*]), which is a known previously reported mutation.6
A higher tendency to develop cerebral neoplasms has been reported.8 A follow-up MRI and clinical evaluation annually is planned.
Usually, L-2-HGA is detected in early childhood, this patient’s adult presentation with a mild phenotype is another example of similar cases reported in the literature.7,8
The therapeutic effect of flavin adenine dinucleotide (FAD) of its precursor, a trial of riboflavin may be considered in the future, with monitoring its impact on disease progression.
In conclusion, physicians should be aware of the early signs of developmental delay and subsequently, the optimum choice of radiological study to perform along with the imaging features in L-2-HGA, which are vital for an early diagnosis of such cases.
Patients with mild phenotype L-2-HGA can remain undiagnosed until adulthood. While developmental delay is almost universal in all patients with L-2-HGA, patients can present without the common features of epilepsy and macrocephaly while manifesting the less common signs, such as dystonia, which might lead to underdiagnoses.
Acknowledgment
We acknowledge the patient and her family members for consenting to participate in this study. The staff of the Radio Imaging and Information Technology Department of our institution was kind to provide details of the patient’s MRI.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received March 3, 2024.
- Accepted April 17, 2024.
- Copyright: © Saudi Medical Journal
This is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.