


Abstract
Objectives: To measure the incidence of vaso-occlusive crises (VOC) and the role of hydroxyurea (HU) in reducing VOC in sickle cell anemia patients being treated at a large tertiary care setting in Kingdom of Saudi Arabia (KSA). The secondary objective of this study is to observe the gradual improvement in laboratory data (white blood cell [WBC], platelets, mean corpuscular volume [MCV], hemoglobin [Hgb], HgbF) following regular use of HU.
Methods: Clinical effectiveness of HU was evaluated in a large pediatric population using a retrospective cohort, non-interventional, pre-post treatment study designed to control disease severity selection bias. The cohort included children with SCA (sickle cell (SS), sickle-beta thalassemia) at King Saud Medical City, Riyadh, KSA, who initiated HU between January 2012 and June 2017. For each patient healthcare utilization, laboratory values, and clinical outcomes were observed for an equal duration of time pre and post hydroxyurea.
Results: Out of 416 SCD patients, 128 children with SCD who initiated HU, of them 82 met the eligibility criteria. After initiation of HU, there was significant reduction in both VOC (80%) and length of stay (LOS) (73%). Significant increase in Hgb (13%), MCV (10%), and HgbF (28%) and significant decrease in WBC (28%) was observed. Only the mean platelet count decreased by 3% with a p>0.05.
Conclusion: Hydroxyurea treatment significantly decreased episodes of VOC and LOS, it also led to reductions in hospitalizations and significant improvement in complete blood count indices.
Sickle cell disease (SCD) is the most common genetic disease, worldwide. The annual number of newborns with SCD was estimated to be 305,800 globally in 2010; by 2050, that number it is expected to increase by approximately one-third to 404,200. Globally, it is estimated that, between 2010 and 2050, the overall number of children born with SCD will be 14,242,000.1,2
Sickle cell disease is an autosomal recessive inherited hemoglobinopathy, it was first described in 1910 by James Herrick.3 The genetic abnormality is due to a substitution of the amino acid valine for glutamic acid at the sixth position on the beta globin chain, which results in the hallmark clinical sequelae of vaso-occlusive phenomena and hemolysis.
Sickle cell disease is a common problem in Kingdom of Saudi Arabia (KSA); it was first reported in 1960 in the Eastern Province.4 A nationwide community-based survey conducted over a period of 2 years, 2004-2005, reported a prevalence of SCD as high as 24/10,000. The prevalence is highest namely, 145/10,000 in the Eastern region (Dammam), followed by the Southern region (Jizan, Najaf, and Baha) with a prevalence of 24/10,000, and then the Western region (Riyadh and Qasim), with a prevalence of 12/10,000; no cases of SCD have yet been reported in the Northern region.4
Over the past few years, the development and use of hydroxyurea (HU) for supportive care and treatment of SCD patients, have significantly improved clinical outcomes for this disease. It was initially approved for use in adults by the United States of America Food and Drug Administration (FDA) in 1998, and, in 2007, the European Medicines Agency authorized HU for pediatric and adult patients with SCA.5
This medication is a ribonucleotide reductase antagonist that increases fetal hemoglobin production and total hemoglobin while reducing intracellular HbS concentration, which, ultimately results in the reduction of polymerization.6 It increases nitric oxide bioavailability while reducing the neutrophil count and intracellular adhesion.7
The safety and efficacy of HU is well documented in the literature on its use in children and adults, although there has been an increase in its use it continues to be under used today.8,9 Many prospective studies have reported significant improvement in hematological parameters as well as a reduction in acute complications, such as painful vaso-occlusive crises (VOC), chest syndrome, blood transfusions, and hospitalization.10-18
Based on the NHLBI guidelines published in 2014, children 9 months and older with SCA should be offered HU at a starting dose of 20 mg/kg/day. Complete blood count (CBC), with a differential and reticulocyte count, should be monitored every 4 weeks to ensure that the absolute neutrophil count is maintained between 2000/µL and 4000/µL and the platelet count is ≥80,000/µL. The dose can be increased by 5 mg/kg every 8 weeks to the maximum tolerated dose or 35 mg/kg/day, based on the clinical or laboratory findings. Once the patient is taking a stable dose, laboratory values can be monitored every 2-3 months.19
A review of 26 articles conducted in the United States of America in February 2008 consisted of one randomized controlled trial, 22 observational studies (11 with overlapping participants), and 3 case reports of adverse events in children with SCD; evidence showed that HU can reduce painful events and hospitalizations in children of all ages with HbSS.20
We found 3 studies on the pediatric population of SCD in the KSA. The first study was conducted in the Eastern Province from 1994-1998; it included pediatric and adult populations, with ages ranging from 10-36 years. That study included a total of 36 patients; 27 of those patients, who had completed 12 months of therapy, were analyzed. Hydroxyurea was found to be effective in decreasing the frequency of VOCs in patients with SCD, and there were no major side effects observed.21
A second study, a retrospective review over a 2-year period (2004-2006) was conducted in Medina, KSA. Ten patients, ranging in age between 5 and15, after HU treatment were observed to experience reduced attacks of painful crises and acute chest syndrome along with a significant increase in mean corpuscular volume (MCV) and hemoglobin F values in a laboratory investigation.22
The third was a cross-sectional study, conducted at King Khalid University Hospital, Riyadh, KSA, between January and March 2014.23 Sample size of 140 people, of which 21 were pediatric patients ranging in age from 11 and 15 years. Patients who strictly complied to HU treatment were observed to suffer less complications and have fewer sickle cell-associated crises.23
Other than the previously mentioned studies, there is very little data on the use of HU in the Saudi pediatric population. Previous research from Europe, South East Asia, and the Americas demonstrate the effectiveness of HU in decreasing painful crises and improving growth in children with SCD, with no documented severe toxicity. However, the benefit of this research has been undermined in KSA, and an investigation of the effectiveness of using HU to treat SCD has not been conducted in a large pediatric population. Thus, it is believed that the present study will contribute to the literature because all reported evidence supports this proposition.
Methods
This retrospective cohort study was conducted in the Hematology Department, King Saud Medical City, Riyadh, KSA. Children with SCD admitted as inpatients and as hematology outpatients, between January 2012 and July 2017, were included.
Patient selection was based on the following criteria: age <14 years who were prescribed HU due to frequent painful crises (more than 2) or one and more acute chest syndrome within a year. Patients who had poor compliance to treatment and whose serum creatinine was >1.0 mg/dl or those with serum alanine transferase (ALT) >2 times the upper limit of normal value were excluded from the study.
All cases were retrieved retrospectively and basic demographic information, including age and gender were recorded. Pre and post-treatment laboratory values for Hb g/dl, MCV, (FL), HbF %, white blood cell, and platelet were recorded and abstracted from each patient chart for an equal duration (namely, minimum 1.5 years pre and post-treatment to max: 5 years pre and post-treatment). In addition to the clinical characteristics, the number of painful crises, the number of acute chest syndrome episodes, and length of stay (LOS) in the hospital due to VOC were also recorded. The collected data was recorded in a specially designed case report form; compliance information was collected from the patients’ parents and the modified Morisky medication adherence scale (MMMAS) was used to assess the patients’ adherence to HU.
Ethical approval was obtained from the International Review Board for the collection and analysis of all data. All the patients were started on HU 15 mg/kg orally (except one who received 10 mg/kg) as a single daily dose; it was increased by 5 mg/kg every 8 weeks in the absence of toxicity. Toxicity of HU was determined when any one of the following was presented: A drop in the absolute neutrophil count (<2000 x 106/L); drop of Hgb more than 20% from the baseline or Hgb decreasing to <5 g/dl; a platelet count decrease to 80.000 × 106L; a 50% increase from baseline of serum creatinine; or alanine aminotransferase increasing to twice the upper limit of normal.
In cases of toxicity, HU was stopped for at least one week. Once the toxicity was resolved, the treatment was resumed at a dose 2.5 mg/kg less than the dose at which the toxicity occurred.
Statistical analysis
Data was analyzed using Statistical Package for Social Sciences for Windows, version 20 (IBM Corp., Armonk, NY, USA). The quantitative variables (age, HB, HgbF, WBC, MCV, platelet count, VOC, LOS) were presented using mean, standard deviation (SD), and the qualitative variable (gender) was presented by using a frequency table and percentage. A paired samples t-test was used to determine the differences in study parameters before and after the initiation of HU. A p<0.05 was considered statistically significant.
Results
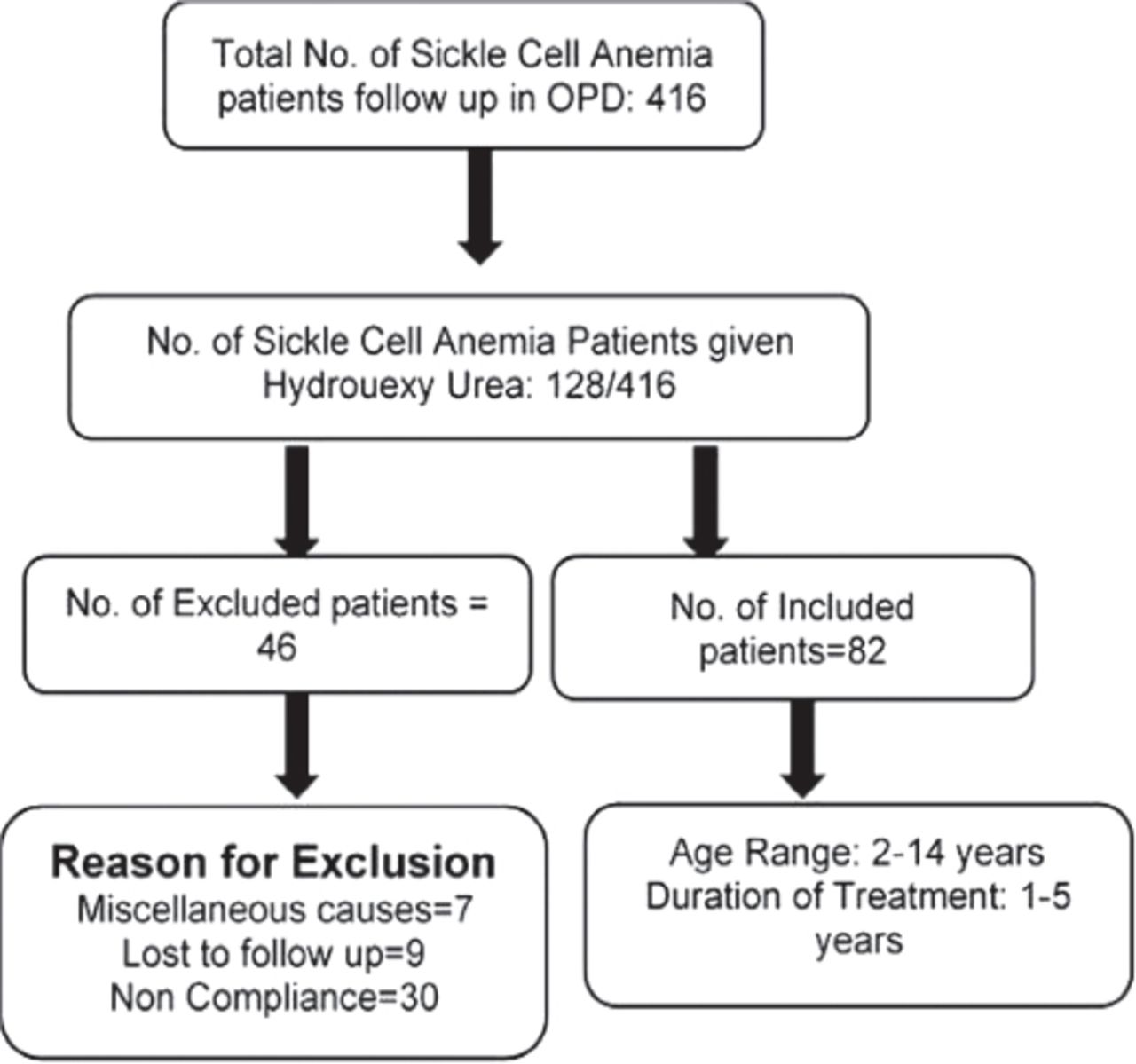
A total of 416 SCD patients were reported; 128 patients fulfilled the criteria and were started on HU. Only 82 of these patients were followed until the end of the study with good compliance; the rest were excluded due to poor compliance or irregular follow-up (Figure 1). Of these patients, 61/82 were SSA while 21/82 were sickle-beta thalassemia (Sthal β°).

Screening and enrollment of participants. OPD: out patient department
Demographic characteristics and hydroxyurea dosage
The mean age of the cases, at the start of our study, was 7.27±3.04 years. Of these, 29 (35.4%) patients were female and 53 (64.7%) were male. The mean dose of HU at the start was 16.18±3.80 mg/kg/day. The mean of the maximum dose at the start was 22.15±3.18 mg/kg/day, and the mean dose of HU at current was 20.90±3.16 mg/kg/day.
Laboratory data before and after initiation of hydroxyurea
Before treatment, the mean Hgb was 8.31±1.42; after treatment it was 9.40±1.51, (p<0.001). The mean HgbF before treatment was 10.07±6.89 and after treatment was 12.93±6.68, (p<0.001). The mean WBC before treatment was 14.53±4.30 and after treatment was 11.31±3.02, (p<0.001). The mean MCV before treatment was 77.52±8.84 and after treatment was 84.18±10.37 (p<0.001). The mean platelet count before treatment was 415.90±135.78; after treatment it was 403.87±120.43, with no significant improvement (p=0.215) (Table 1).
Descriptive statistics before and after hydroxyurea.
Vaso-occlusive crisis and length of stay
Before treatment, the mean VOC was 5.98±2.21; after treatment it was 1.76±1.41, indicating significant improvement (p<0.001). The mean LOS before treatment was 31.27±17.58; after treatment it was 8.50±9.74 (p<0.001).
Discussion
This study demonstrated that using HU to treat SCD (SSA and Sthal β°) has definitive clinical and hematological benefits. The results showed that administration of HU significantly reduced VOCs by 80% and decreased the LOS by 73%. It was also observed that Hgb improved by 13%, white blood cell (WBC) decreased by 28%, MCV improved by 10%, and HgbF improved by 28%, platelet count was an exception since it decreased by 3%. Similar results were found by Lopes de Castro Lobo et al,24 in a group of 1760 children ranging in age from 3-18 years; of the 267 patients treated with HU Hgb, MCV, and HgbF increased while WBC and platelet count decreased within a 12-month period. A significant decrease in the rate of hospitalizations, the duration of hospitalizations, and the number of ER visits was also observed. A higher survival rate for children treated with HU, even though they had a more severe version of SCD, in comparison to those that had not been given HU, was also noticed.25
Previously, comparable findings were reported by Sharef et al,26 in a population of 142 Omani children ranging in age from 2-16 years, and by Phillips et al,27 in a population of 37 British children with a mean age of 7 years; in both these cases the Hgb, MCV, and HgbF were found to be significantly increased in as short as 6 months after the initial dosage. While dosage escalation is required to maximize the beneficial effects of HU, the advantages of using this medication can be observed within 50 days after its initial administration.27,28
In 2012, Jain et al,38 conducted a trail of low-dose HU in an Indian population of 60 children ranging in age from 5-18 years in comparison to a placebo treatment; the Hgb and HgbF both increased and were significantly higher in the HU group.27
Quarmyne et al,28 conducted a large retrospective population-based study between 2009 and 2011; they found similar results regarding VOC, LOS, Hgb, WBC, MCV, and HgbF counts in a population of 134 American children (median age=7.5 years). They found that after HU initiation, children younger than 7 years of age had an even greater reduction in the rate of VOCs and LOSs in comparison to older children.28
Hemoglobin F is the most important predictor of clinical severity in SCD patients. A low level of HgbF is linked with a higher risk for VOC, organ damage, and early mortality.29 An increase in HgbF is linked with a decrease in the clinical severity of SCD, leading to less need for transfusions, VOCs, and hospitalizations.30 Since 1990, HU has been a promising pharmacological therapy for SCD due to its ability to induce HgbF.31 Silva-Pinto et al,32 argued that the mechanisms with which HU works in SCD cases are still only partially known, and clinical improvements started before HgbF increment, which includes a reduction in sickle adhesiveness facilitated by a reduction of surface adhesive molecules, reduced platelet and reticulocyte counts, induction of nitric oxide production, an increase in the cell volume of sickle erythrocytes, and a decrease in WBCs.32 In SCD patients, an elevated WBC count is linked with morbidity and mortality, so it is important that the WBC of patients with SCD does not surpass the normal limit.33
In our study, we observed a drastic reduction in the rates of VOC (80%) and LOS (73%). As previously reported by Ferster et al,34 Jain et al,29 and various other studies, VOC was the most common complication and indication for hospital admission. After administration of HU admissions and durations of admission fell significantly, regardless of whether patients were administered a high or low dose of HU.25,26,38
The decrease in platelet count observed in our study was less than 3%, which is in line with the results reported by Wang et al,10 who also observed a non-significant reduction in platelet count after administration of HU. However, Sharef et al,25 observed a significant reduction in platelets in both the low and high dose HU groups.
Hydroxyurea is an ideal therapeutic agent for use in children with SCD. It requires only one daily dose, it has excellent bio-availability after oral administration, and it can be consumed in the form of tablets or stirred in water.32 It remains greatly under-utilized for SCD in children, primarily due to provider inexperience and the safety concerns expressed by parents. Hydroxyurea has been found to have predictable toxicities that are dose-dependent, minimal and reversible and few, if any, immediate side effects.25,26,30,32 In young patients, the immediate side effects of HU have been reported to be mild and tolerable.29 Within our participants, the dosage of 2 female patients could not be escalated due to the concerns of their respective mothers of mild hair loss, which on clinical examination was not apparent. Another side effect was seen when 3 male patients developed neutropenia in the range of 900-1200, due to their HU dosage, which was then put on hold for 2 weeks to reverse this effect. Two of these 3 male patients had a history of upper respiratory tract infection (URTI) one week prior. During a follow-up after the 2-week medication hold, the count returned to normal and HU was resumed at a lower dosage of 2.5 mg/kg less than the previous dose. There were no more similar episodes after this, even with the increase in dose. Two other male patients complained of dyspepsia, secondary to the HU, which was controlled by an oral antacid.
Another reason HU remains under-utilized is because of an unexplained reluctance to consider SCD a life-threatening disease meriting immediate and aggressive preventive therapy, and not just symptomatic management. Due to this reluctance many people affected by SCD are less likely to receive treatment until an acute complication or serious form of organ damage occurs.31
Wang et al,10 considered a 3-year duration of observing children on HU to be a short time frame. Our study had the advantage of being able to observe some children for as long as 5 years; however, some of the children were observed for as few as 1.5 years. The ages of the children included in this study population also varied vary greatly; while the average age was 7.27 years, the youngest child was 2 and the oldest was 14.
Study limitation
A strong limitation of our study is that the patient population included a wide range of local citizens who had access to the tertiary center and treatment; therefore, it would be difficult to extrapolate these findings to different ethnicities. A noteworthy limitation of this study was that it was retrospective in nature and was a single center study.
While, to date, there is more than 15 years of data on exposure among adults and more than 12 years of data on exposure among children, thus far, continued vigilance is warranted, especially in regard to the effects HU may have on growth and pubertal development in young children.32
In conclusion, due to the large scale of the local Saudi population used, we can sufficiently deduce that the use of HU is efficient in reducing complications associated with SCA in the pediatric population. Based on the study’s findings, it can be concluded that HU treatment can significantly reduce the number of hospitalizations and the LOS due to VOCs with significant improvements in the CBC indices. Based on the discussion presented above, it can be said that HU fulfills the criteria of an ideal therapeutic agent for treating SCD patients. It has a significant impact on hematological parameters as well as clinical efficacy in terms of VOC and hospitalization; moreover, HU is safe, effective, and has minimal short-term side effects. It can be effectively used in younger patients before the development of long-term complications and end organ damage. However, the long-term side effects of HU require further observation.
Further studies are recommended on the long terms side effects of HU on the Saudi pediatric population, such as end stage organ failure and potential reduction in sperm count. Puberty has been observed to be delayed in children who suffer from SCA, the extent to which the onset of puberty is improved after the use of HU in the Saudi pediatric population should be observed.
Acknowledgment
The authors gratefully acknowledge Scribendi-Editing and Proofreading Services for English Documents (www.scribendi.com) for English language editing.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received July 15, 2019.
- Accepted October 29, 2019.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.