


Abstract
Glutaric aciduria type 1 (GA1) is an inherited inborn error of metabolism caused by a deficiency of the enzyme glutaryl Co-A dehydrogenase (GCDH). Here, we report a 14-month-old Saudi boy with GA1 who presented with severe dystonia and was mis-diagnosed as cerebral palsy (CP). He presented to our institute with encephalopathy following an episode of gastroenteritis. His physical examination showed dystonia and spastic quadriplegia. His investigations revealed elevated both urinary 3-hydroxy glutaric acid, and serum glutarylcarnitine. The DNA analysis confirmed homozygosity for a mutation in the GCDH-coding gene (c.482G>A;p.R161Q). This case alerts pediatricians to consider GA1 as a differential diagnosis of children presenting with dystonic CP.
Glutaric aciduria type 1 (GA1) (OMIM #231670) is an inherited autosomal recessive metabolic disorder caused by mutation in the glutaryl Co-A dehydrogenase (GCDH) gene.1 This gene maps to chromosome 19p13.2. If mutated, it results in deficiency of the enzyme GCDH.1 This mitochondrial enzyme is involved in the metabolism of lysine, hydroxylysine, and tryptophan.2 Its deficiency lead to accumulation of glutaric and 3-hydroxyglutaric acids, which are toxic to the brain and cause striatal injury. The prevalence of GA1 is estimated to be one in 100,000 newborns.3 Affected individuals in infancy, may initially present with macrocephaly, but otherwise, they are neurologically healthy. Encephalopathy is usually triggered by intercurrent infection, such as acute gastroenteritis, which affects most untreated patients.4 It may progress to severe dystonia, choreoathetosis, and spastic quadriplegia if left untreated.4 When presented clinically, GA1 is usually diagnosed by measuring serum glutarylcarnitine, urinary excretion of glutaric, and 3-hydroxyglutaric acid.2 To prevent permanent and irreversible neurological damage, screening for GA1 has been included in expanded neonatal screening programs in many countries.3,4 A positive glutarylcarnitine blood spot-screening test requires assessment of urine organic acids.2 The diagnosis of GA1 is confirmed by measuring GCDH enzyme activity, or performing mutation analysis.4 Different mutations have been reported in the GCDH gene.5,6 The aim of management in GA1 is to prevent neurological complications, including movement disorders and seizures. Treatment consists of low lysine diet with L-carnitine supplementation. Favorable outcome was reported in patients who were diagnosed and started treatment early.3 We report a Saudi child with GA1 who presented with dystonia and misdiagnosed as cerebral palsy (CP) to alert pediatricians to consider GA1 as a differential diagnosis in patients with dystonic cerebral palsy, as early intervention will prevent permanent neurological damage.
Case Report
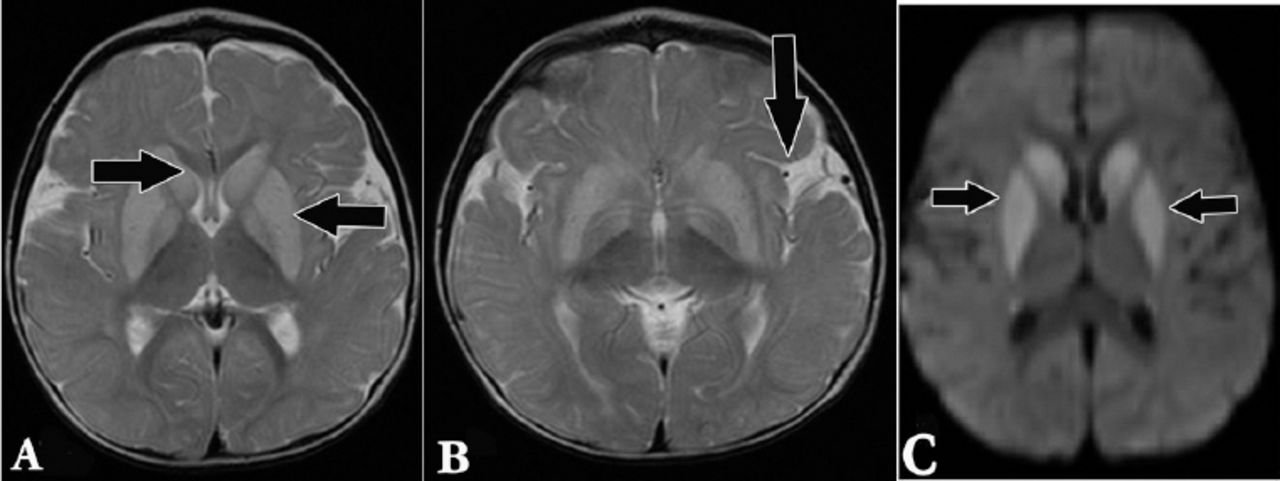
A 14-month-old boy presented to our hospital with developmental regression and severe dystonia. He was born at term by spontaneous vaginal delivery following an uneventful pregnancy with no history of perinatal asphyxia. He is the first baby of first-cousin parents with no family history of metabolic disorders or early neonatal deaths (Figure 1). He developed neonatal jaundice on the second day of life, and was treated with phototherapy for 5 days. He was noted to be floppy since early infancy with delayed gross and fine motor development. He achieved head control after the age of 8 months, and started to sit unsupported at 10 months of age. However, he lost some of the developmental skills that he had gained after an acute gastroenteritis at the age of 11 months. This acute illness was complicated by encephalopathy and seizures. Extensive workup was carried out including cerebrospinal fluid analysis that was negative for viral and bacterial infections. He remained with severe spasticity, not responding to extensive physiotherapy and Baclofen therapy. His initial evaluation at our hospital showed a spastic child with severe dystonic posture and failure to thrive. His investigations revealed normal serum lactate and ammonia with no evidence of metabolic acidosis. His brain magnetic resonance imaging (MRI) showed abnormal high signal intensity at basal ganglia and widened Sylvian fissure (Figures 2 & 3). Based on clinical presentation along with the MRI findings, the diagnosis of GA1 was considered. He was started on intravenous dextrose 10%, oral carnitine, and low protein diet. Few days later, his urine organic acids result showed elevated 3-hydroxyglutaric acid, while serum amino acids were normal. Acylcarnitine profile revealed high glutarylcarnitine, consistent with the diagnosis of GA1. The deoxyribonucleic acid extracted from lymphocytes was used to amplify the 11 coding exons, and the corresponding flanking sequences of the GCDH gene. The polymerase chain reaction products were analyzed by sequencing in both forward and reverse directions. Sequence analysis identified 2 copies of a missense mutation, c.482G>A in the coding region of the GCDH gene. This homozygous mutation predicts an amino acid change of arginine (R), glutamine (Q) at codon 161 of the dehydrogenase protein (p.R161Q).6 Also, both parents were found to be carriers of the same mutation. This mutation has been previously reported in patients with GA.6 Our patient was maintained on low lysine diet with supplementation of L-carnitine 100 mg/kg/day. His follow-up showed complete resolution of the extrapyramidal signs with significant improvement in motor skills and cognition.
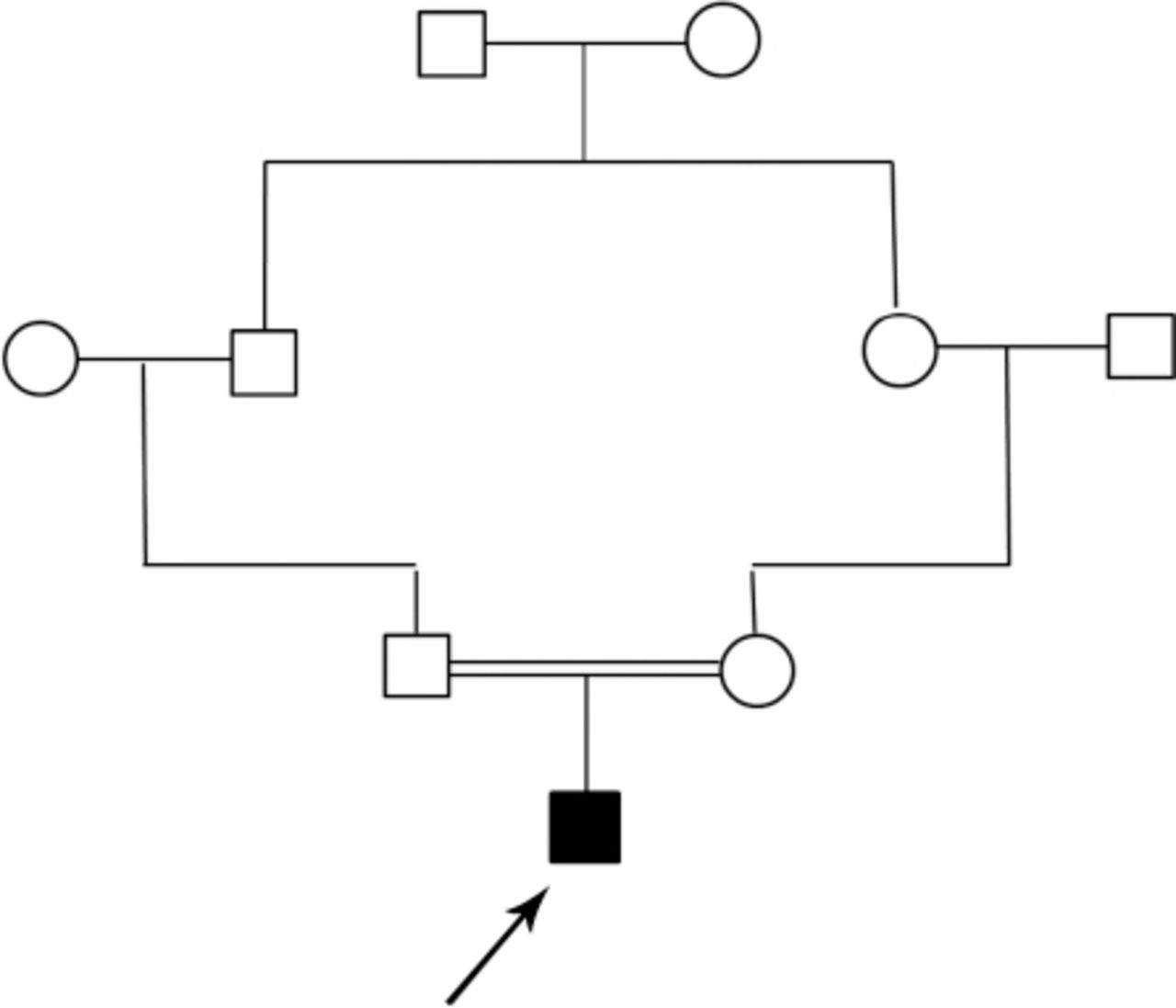
Family pedigree of a 14-month-old boy with glutaric aciduria type 1. ◻ - Male, ◯ - female, ◼ - affected.

A magnetic resonance imaging of the brain, axial T2 weighted image showing: A) abnormal high signal intensity at basal ganglia; B) widened Sylvian fissure; and C) axial diffusion weighted imaging showing diffusion restriction at the basal ganglia.

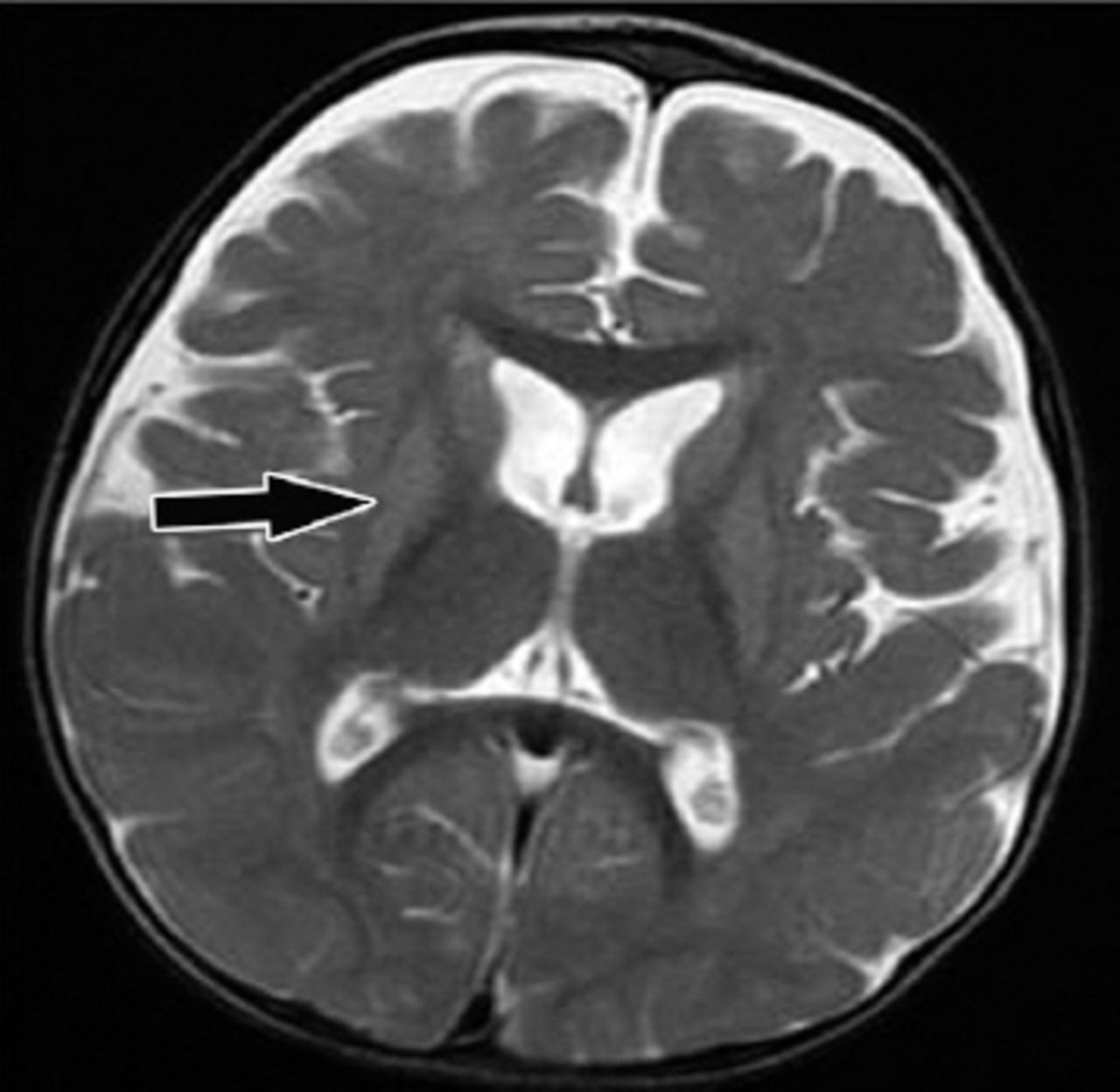
Follow up magnetic resonance imaging brain study after 3 months. Axial T2 weighted image showing reduced size (atrophy) of basal ganglia and progressive bilateral fronto-temporal atrophy.
Discussion
Glutaric aciduria type 1 is a rare metabolic disorder caused by deficiency of the enzyme GCDH that is involved in the metabolism of lysine, hydroxylysine, and tryptophan.1 The prevalence of GA1 in the Kingdom of Saudi Arabia (KSA) is unknown; however, it is expected to be higher than the global figure due to the high incidence of consanguinity in this country.7 The recently introduced universal neonatal screening program in KSA included GA1.8 This program is expected to reveal the magnitude of this disease together with other inherited diseases included in the screening panel. Furthermore, newborn screening for GA1 identifies affected infants before the development of neurological signs, and thus, can prevent associated morbidity and mortality.3,4 Similarly, recent data1,3,4 from the expanded neonatal screening indicated that patients who were diagnosed by screening have better prognosis in comparison with those who were diagnosed clinically. Unfortunately, our patient was not detected on neonatal screening, as it was not available in the hospital where he was born. On the contrary, he presented with severe dystonia and encephalopathy picture following acute gastroenteritis. This agrees with the previous reports that recognized acute encephalopathic crisis in infancy is often precipitated by infection, vaccination, surgery, or trauma.1,3
Our patient was misdiagnosed initially as dystonic CP. In contrast to the progressive dystonia resulting from GA1, CP is a non-progressive motor disorder affecting the developing fetal or infant brain.9 The CP affects approximately 2-2.5 per 1000 live births.9 It is usually caused by hypoxic ischemic encephalopathy secondary to perinatal insults. Therefore, it presents in the first half of the first year with developmental delay and neurological signs. In contrast, GA1 usually presents in the second half of the first year of life with regression of milestones following an intercurrent infection. Thus, careful history taking and thorough clinical examination can differentiate between dystonia caused by GA1 and CP.9
Brain magnetic resonance imaging is an important diagnostic tool for GA1, and the findings may be highly suggestive of this diagnosis. The brain MRI findings of our patient revealed abnormal high signal intensity in the basal ganglia and widened Sylvia fissure that alerted us to the possibility of GA1. Similar MRI findings were reported in the literature in patients with GA1.10 The diagnosis of GA1 was confirmed in our patients based on the presence of elevated urinary 3-hydroxyglutaric acid, high serum glutarylcarnitine, and identification of the homozygous nonsense mutation (c.482G>A;p.R161Q) in GCDH gene. Different mutations, including the one identified in our patient had been reported previously in different ethnic groups.5,6 Aggressive treatment of the intercurrent infections of patients with GA1 is important in order to prevent complications. The goal is to prevent catabolism by providing high-energy intake with an extra 20% of caloric requirements through carbohydrates and lipids. Also, cessation or reduction of protein intake to 50% or less, depending on the severity of the illness, and doubling the dose of carnitine is recommended.
In conclusion, this case report alerts pediatricians to consider GA1 as an important differential diagnosis of dystonic CP. Early treatment of GA1 prevents long-term neurologic disabilities. We would like to emphasize the importance of the newborn screening to detect GA1 patients before symptoms occur. Also, all children with neurologic symptoms of unknown origin such as CP, should certainly undergo work-up for inborn errors of metabolism.
New Peer Reviewers
Join our team of expert peer reviewers for Saudi Medical Journal by registering through the website at http://www.smj.org.sa/_Authors/ and select “register now” or sending an enquiry and summarized CV to smjadmin{at}psmmc.med.sa. Note that SMJ reviewers, whose reviews are returned on time and are judged satisfactory by the Editors, may receive 1 CME credit per review, with a maximum of 5 credit per year, from the Saudi Council for Health Specialties.
Footnotes
Disclosure. This study was supported by the College of Medicine Research Center, Deanship of Scientific Research, King Saud University, Riyadh, Kingdom of Saudi Arabia.
- Received April 16, 2015.
- Accepted July 12, 2015.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.




{kind=link}
{kind=link}
{kind=link}