


Abstract
We describe a 10-year-old boy with a rare large multicystic pulmonary chondroid hamartoma in the right lower lobe presenting with severe respiratory distress. The radiological evaluation showed a large multicystic lesion in the right lower lobe with pneumothorax. Commonly, pulmonary hamartomas are asymptomatic, small in size, and are diagnosed incidentally in adults. Our case is highly unusual due to its young age, prominent clinical symptoms of severe respiratory distress with pneumothorax at presentation, very large size, and prominent cystic change. The lesion was surgically excised, and histopathological features were compatible with a multicystic chondroid hamartoma.
Hamartomas are an abnormal proliferation of a mixture of tissue elements that are normally present in that organ. Pulmonary hamartomas (PH), also known as chondroid hamartomas, are the most common benign tumors of the lung in adults.1 They are slow growing, non-infiltrative, and nodular. Histologically, they show a mixture of mature mesenchymal tissue like adipose tissue, cartilage, bone, smooth muscle bundles, and fibromyxoid tissue in varying proportions.2 In addition, cleft-like spaces lined by respiratory epithelium are common. Males are 4 times more affected than females. The PHs are common in the fifth and sixth decade of life, and they tend to occur in the peripheral lung fields. Radiologically, they are typically described as coin lesions due to their smooth, sharply demarcated outline, and round shape. Identification of ‘popcorn-like’ calcification on plain radiograph is diagnostic when present. The PHs are mostly asymptomatic, diagnosed incidentally, measuring less than 4 cms in diameter, and they rarely show extensive cyst formation. We report a case of a large multilocular cystic chondroid pulmonary hamartoma in a 10-year-old boy. Our objective in presenting this particular case is to highlight the highly unusual pediatric age group, unique clinical and radiological findings, prominent cystic change, and very large size.
Case Report
A 10-year-old otherwise, healthy boy presented with 4 weeks history of cough and progressive severe shortness of breath with white colored sputum. There was no wheezing, fever, or chills. Physical examination revealed a reduced air-entry on the right side of the chest. Chest radiography showed a right-sided pneumothorax with right lung lobe radiopaque mass. He was immediately started on intravenous fluids (IV) and antibiotics, inhaled bronchodilators, and a chest tube was inserted. A chest CT scan with IV contrast was ordered, and it showed a large multi-septated cystic lesion in the right lower lobe with compression and atelectatic changes, mild mediastinal shift to the left, and significant pneumothorax with a small amount of pleural fluid. Some of the cystic spaces were air filled, while others were fluid filled (Figure 1). The left lung, heart, and great vessels in the mediastinum were normal. A clinical diagnosis of a complicated congenital pulmonary airway malformation (CPAM) with moderate pneumothorax was made. He underwent a right-sided thoracotomy. On opening, a large cystic mass with fibrous adhesions to the diaphragm, the right middle lobe, and right lower lobe of the lung, was identified. The mass was released from the surrounding structures and excised/enucleated. The background lung tissue was not resected. The mass was sent for histopathological evaluation. Gross pathological examination showed a well-circumscribed, smooth, cystic mass measuring 11.3 × 10.5 × 5.0 centimeters and weighing 178 grams. The cut-surface showed multiple, variably sized cysts filled with clear fluid. The walls of the cysts appeared bumpy and nodular. Microscopically, the cysts were lined by cuboidal to flattened cells. Numerous solid micromodules were seen in the cyst wall. The nodules consisted of benign mature adipose tissue, fibrous tissue, and abundant mature cartilage (Figure 2). A diagnosis of a multilocular cystic chondroid pulmonary hamartoma was made. The respiratory symptoms disappeared postoperatively. A repeat CT scan was performed one month post-surgery, and it showed a residual small pneumothorax. No mass was identified.
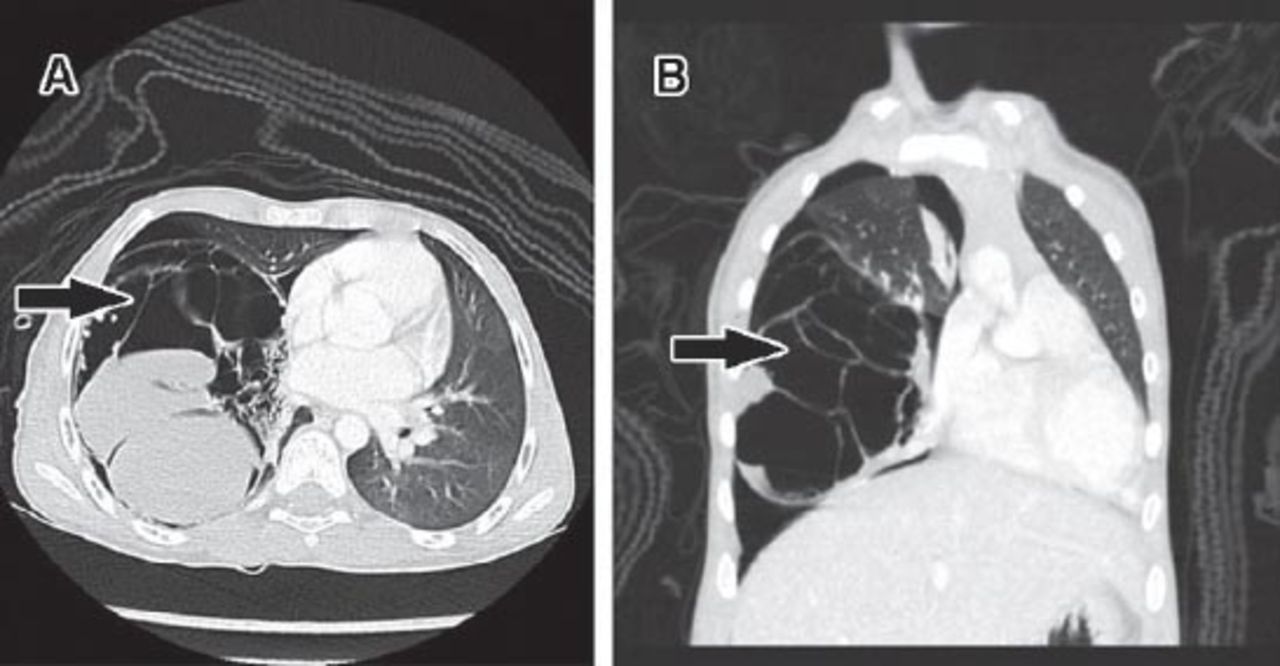
Computed tomography scan of the multicystic pulmonary hamartoma showing: A) Axial cut demonstrates a large multiseptate cystic lesion in the right hemithorax (arrow), some of the cysts are air filled while others are fluid filled. This lesion shows mass effect upon adjacent mediastinum. B) Coronal image shows the multilocular nature of this cystic lesion (arrow), and its effect upon the adjacent lung tissue and the pneumothorax.
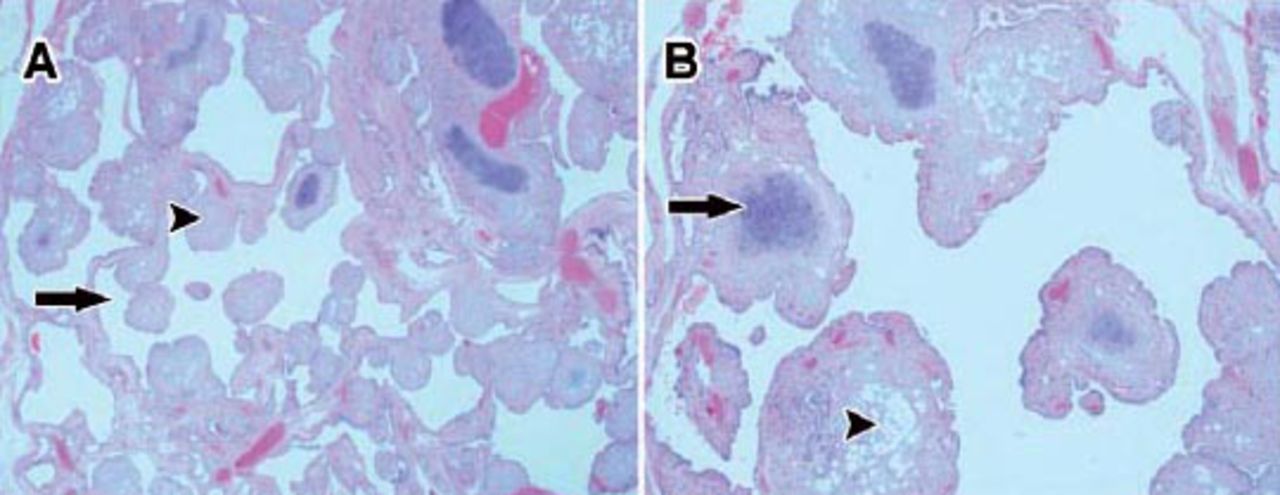
Photomicrograph of the multicystic pulmonary hamartoma showing: A) A mass composed of numerous microcystic spaces (arrow) with multiple micronodules (arrowhead) in the wall of the microcysts. (Hematoxylin and Eosin stain; original magnification ×20). B) The micronodules are lined by benign flattened to cuboidal epithelial cells. The micronodules are composed of hyaline cartilage (arrow), fibrous tissue, and adipose tissue (arrowhead). There is no pleomorphism, abnormal mitosis or necrosis in the any of the cartilaginous, fibrous, or adipocytic mesenchymal elements (Hematoxylin and Eosin stain; original magnification ×40).
Discussion
Hamartoma was first described in the early 1900s.2 It is a common benign tumor of the adult lung seen in the fifth to sixth decade of life, with an incidence of 0.025-0.32%.1 Benign lung tumors are rare in the pediatric age group, of which the relatively common ones are inflammatory pseudotumors, hemangiomas, and adenomas. Pediatric PHs are very rare. The PHs can be parenchymal or endobronchial in location. Parenchymal hamartomas are more common, mostly incidental, and asymptomatic while endobronchial tend to be symptomatic with cough, hemoptysis, and obstructive pneumonia. Awareness of this lesion is necessary as it is a mimicker of primary and metastatic lung tumors, both clinically and radiologically. A CT-guided fine needle aspiration with cytological assessment helps exclude malignancies and other pathologies. Many times PHs are surgically excised either by enucleation or wedge resection, and are diagnosed on histopathology.2 The PHs grow slowly at a rate of 3.2 ± 2.6 mm per year; however, they can double in diameter over time.3 Giant PHs are rare, and most of them have been reported in adults, some as big as 26 cm.4-6 Pediatrics cases of this size have rarely been reported. Ozbudak et al3 reported an 11-year-old with a cystic chondroid PH measuring 18 cm across. Our case measures 11.3 cm in maximum dimension. An endobronchial PH was reported in an 8-month-old infant.7 Our case showed prominent cystic changes. The reason behind the cyst formation is still not clear. Some authors suggest that as hamartomas grow in size, their tendency to become cystic increases. The degenerative changes coupled with the growth leads to formation of cleft-like spaces that ultimately expand into cysts.3 Alternatively, the proximity of a hamartoma to a bronchiole with air-entry and a check-valve mechanism may lead to the expansion of epithelial-lined tubules into resultant cysts.3 We were unable to demonstrate any such connection to any bronchiole in our case.
Initially, hamartomas were considered to be a developmental anomaly, but cytogenetic analyses have shown mutations of high mobility proteins along chromosomal bands 6p21 and 12q13-15, thereby allotting hamartomas the status of a true neoplasm.8 Hamartomas originate from the undifferentiated mesenchymal tissue, which in the case of the lung is the fibrous connective tissue in the bronchial wall. The differential diagnoses for PHs include chondroma, well-differentiated chondrosarcoma, fibroma, and lipoma, all of which are easily excluded by histopathological examination. In our case, due to the age, large size, and multicystic nature, a preliminary diagnosis of CPAMs was made on gross pathological evaluation. In CPAM, an entire lobe of the lung may be replaced by dysfunctional abnormal cysts. A possibility of pulmonary sequestration was also considered. Chondroid cystic malformation associated with trisomy 8 karyotypic anomaly is another close differential diagnosis. Diffuse chondroid malformation was ruled out by the absence of the mesenchymal and adipose tissue typically seen in PHs. In addition, diffuse chondroid malformation has a classical peripheral distribution of cartilage and they tend to be more diffuse. Malignant carcinomatous or sarcomatous transformation of PHs is rare. Complications of large PHs include hemothorax, and pneumothorax.9 Aspergillus colonization of these cysts has also been reported.10
In conclusion, we recommend that despite the rarity of PHs in children, this neoplasm should be considered in the differential diagnosis of any child with a large pulmonary mass, especially since it is benign and surgery is curative. Currently, our patient is being followed up regularly and is doing well.
Acknowledgment
The authors acknowledge Dr. Sami A. Al-Nassar, Department of Thoracic Surgery for his surgical supervision in this case. Also, Dr. Ahmad Amer Al Boukai, Department of Radiology, College of Medicine and Khalid University Hospital, King Saud University, Riyadh, Kingdom of Saudi Arabia, for reviewing the radiology images.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received September 9, 2014.
- Accepted January 21, 2015.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.