


Abstract
The aim of this report is to analyze the clinical features, and mutations of the methylmalonyl CoA mutase (MUT) gene in 2 patients with methylmalonic aciduria (MMA) attending King Saud University Medical City, Riyadh, Saudi Arabia in January 2014. The infants aged 6 days (patient 1) and 3 months (patient 2) with sepsis-like picture, metabolic acidosis, and hyperammonemia were presented. Investigations revealed high propionylcarnitine (C3), elevated urinary methylmalonic acids, 3-hydroxypropionic acids and methylcitrate, consistent with MMA. Sanger-sequencing detected a homozygous novel mutation (c.329A>G; p.Y110C) in the MUT gene in patient 1 and a heterozygous in parents. This mutation is predicted to have a damaging effect on the protein structure and function. In patient 2, we detected a novel homozygous nonsense mutation (c.2200C>T; p.Q734X) and a heterozygous in parents. This mutation leads to a premature stop-codon at codon 734 of the MUT gene. We identified 2 novel mutations in the MUT gene causing isolated MMA.
Methylmalonic aciduria (MMA) is a genetically heterogenous disorder caused by an inborn error of methylmalonate and cobalamin metabolism.1 The estimated prevalence of this condition is one in 50,000 to 100,000 people.2 Most isolated MMA cases are caused by mutations in the methylmalonyl CoA mutase (MUT) gene (OMIM 251000). There are other genes involved in the pathogenesis of isolated MMA, such as MMAA, MMAB, MMADHC, and MCEE.2 The MUT gene encodes the enzyme MUT that converts L-methylmalonyl-CoA to succinyl-CoA. Mutations in the MUT gene alter this enzyme’s structure, or reduce its amount leading to partial or complete deficiency.3,4 As a result, methylmalonyl acid, and other potentially toxic compounds can accumulate in the tissues, causing signs and symptoms of MMA.2 In contrast, patients diagnosed on newborn screening based on the detection of elevated concentration of blood propionylcarnitine (C3) are usually asymptomatic at the time of diagnosis.5 The MMA usually presents with variable severity, from mild to life-threatening during the neonatal period. Affected infants may experience poor feeding, vomiting, dehydration, seizures, hepatomegaly, and delayed milestones.1 Without treatment, this disorder can lead to encephalopathy, coma, and death in some cases.1 Long-term complications of MMA include feeding problems and growth failure, chronic renal disease, pancreatitis, movement disorders, and intellectual disability.5 As molecular studies of MMA in Saudi Arabia are limited, our objective was to analyze the clinical features and novel mutations of MUT gene in 2 patients with isolated MMA attending King Saud University Medical City (KSUMC) in Riyadh, Saudi Arabia in January 2014.
Case Report
Patient 1
A 6-day-old baby boy presented with decreased activity and poor feeding. He was delivered at term by an elective cesarean section due to breech presentation with a birth weight of 3.07 kg. His parents are first cousins with no family history of metabolic or genetic diseases (Figure 1). His initial evaluation showed a lethargic, sick looking baby with severe dehydration. The rest of the systemic examination was unremarkable. He received fluid resuscitation, covered with broad-spectrum antibiotics, and electively intubated and ventilated. His baseline investigations showed hypoglycemia, increased anion gap metabolic acidosis, ketonuria, and serum ammonia of 633 µmol/l (normal value [NV] <80 µmol/l). The working diagnosis was organic acidemia and accordingly our patient was commenced on ammonia scavenger medications, namely, intravenous sodium benzoate and sodium phenylbutyrate, in addition to intravenous arginine hydrochloride, oral carnitine, and hydroxocobalamin intramuscular injections. Four hours later, ammonia was increased to 917 umol/l and hence, continuous renal replacement therapy was started, and the ammonia gradually decreased to reach 298 in less than 24 hours. However, on the second day of admission our patient deteriorated, became hemodynamically unstable despite being on maximum inotropic support. He developed severe coagulopathy and died of presumed sepsis while the ammonia level was below 300 umol/l. The result of urine organic acids was consistent with MMA. It showed highly elevated methylmalonic acids. It showed highly elevated 3-hydroxypropionic acids and methylcitrate, while acylcarnitine profile showed propionylcarnitine of 21 Um (cutoff 10 uM), and the ratio of propionyl carnitine to acylcarnitine (C3/C2) was 2.14 (cutoff 0.4) (Table 1). In addition, high glycine was observed in serum amino acid.
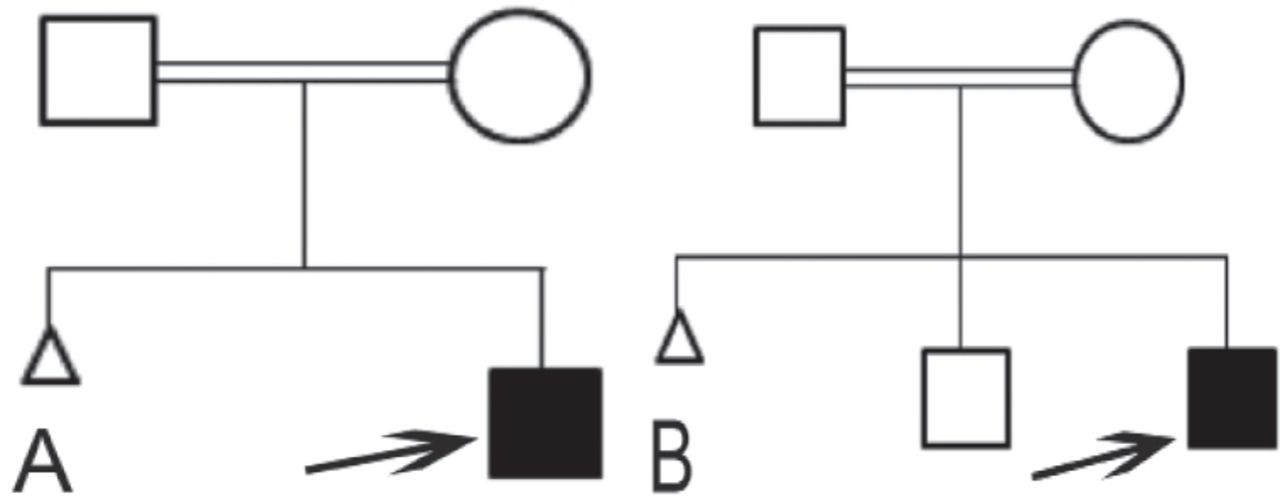
Family pedigree of A) patient 1 and B) patient 2. ◻ - unaffected male, ◯- unaffected female, ◼ - affected male ⧍ - miscarriage
Characteristics of patients with methylmalonic aciduria.
Patient 2
A 3-month-old boy presented with sepsis-like picture. He was born at term by spontaneous vaginal delivery with unremarkable pregnancy and neonatal history. His parents are first cousins with no family history of inherited disease, or early neonatal deaths (Figure 1). He was well until the age of 2 months when he was admitted to the hospital with respiratory distress treated for pneumonia for 5 days. Ten days later, he presented with fever, poor feeding, and decreased activity. Upon arrival at the hospital, he was sick, looking pale, and tachycardic. The rest of his systemic examination was normal apart from increased tone in the upper and lower limbs. All sepsis screening tests were negative including blood, urine, and cerebrospinal fluid cultures and viral serology. His initial blood gas showed increased anion gap metabolic acidosis and serum ammonia was 161 umol/l and serum methylmalonic acid was 275.2 (NV: <0.4 µmol/L). This biochemical profile was suggestive of methylmalonic aciduria that was also supported by the high urinary methylmalonic acid, elevated 3-hydroxypropionic acids and methylcitrate (Table 1). In addition, acylcarnitine showed high propionylcarnitine and high propionyl carnitine to acylcarnitine ratio. Our patient responded nicely to low protein formula, oral carnitine, and hydroxocobalamin intramuscular injections. His ammonia was maintained within the normal limits, and his growth and development were age-appropriate.
Genetic findings
Blood samples (5 mL) were collected in EDTA tubes from patient 1 and patient 2. The tubes were centrifuged at 5,500×g for 5 min. The DNA was extracted from the buffy layer using the illustrated blood genomic Prep Mini Spin kit (GE Healthcare, Buckinghamshire, UK) and stored at -20°C in aliquots until further use. The DNA samples from both patients were tested for mutations in MUT gene by polymerase chain reaction and direct Sanger-sequencing. Full coding region covering all 13 exons and flanking region were amplified using previously described primers (Table 2).3 Successfully amplified fragments were sequenced in both directions using the M13 forward and reverse primers and the BigDye terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA). Fragments were electrophoresed on the 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA) according to the manufacturer protocol. All the sequenced fragments were then analyzed using SeqScape software v2.6 (Applied Biosystems, Foster City, CA). Prediction of potential pathogenic effect of the detected mutation(s) were performed utilizing SIFT (http://sift.jcvi.org/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/).6,7
Primers used for polymerase chain reaction and sequencing of the methylmalonyl CoA mutase gene.3
In patient 1, we detected a homozygous mutation (c.329A>G;p.Y110C) and heterozygous in both his parents. This mutation is novel (not reported previously). To study the effect of this mutation on the protein structure and/or function, we utilized SIFT (http://sift.jcvi.org/) and PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), both databases predicted that this mutation will have an effect on the protein structure and function.6,7 The SIFT predicted it to be not tolerated, and PolyPhen2 predicted that this mutation would be possibly damaging. This mutation was not detected in 100 chromosomes of matching ethnicity.
In patient 2, we detected a homozygous nonsense mutation (c.2200C>T;p.Q734X) in the patient and heterozygous in both his parents. The mutation will lead to a premature stop-codon (X) at codon 734 of the MUT gene. This mutation is novel (not reported previously) and was not detected in 100 chromosomes of matching ethnicity
Discussion
Isolated methylmalonic aciduria is an autosomal recessive disorder caused mainly by a deficiency of the enzyme MUT.1 It usually presents with non-specific symptoms in infancy.2 In this report, we described 2 infants with severe isolated MMA presented early in infancy with sepsis-like picture, metabolic acidosis, and hyperammonemia. While the second patient responded to the therapeutic measures including intravenous fluid, carnitine, and low protein formula, the first patient died from presumed septic shock. Patients with MMA identified by neonatal screening benefit from early treatment that leads to the prevention of acute complications and death in infancy. However, the long-term outcome of screened patients is less well defined as data is scarce.5 Although newborn national screening for genetic diseases operates in most hospitals in Saudi Arabia, it is not available in the hospital were our patients were born. Furthermore, the early and severe presentation combined with the location of the mutation (middle of the MUT gene active-site) suggests our patients would likely be unresponsive to even appropriate early treatment. The clinical presentation of our 2 patients and the abnormal findings on amino acids, acylcarnitine profile, and organic acid were highly suggestive of MMA. We therefore opted to perform mutation study of the MUT gene. We sequenced the full coding regions and splice junctions of this gene in our 2 patients, and detected 2 homozygous mutations. After an extensive search of the literature and available database, we concluded that the 2 mutations described here are novel (not reported previously).
There are 4 other genes where mutations can cause MMA. The mutation rates for genes involved in MMA phenotype are: 60% for MUT; 25% for MMAA; 12% for MMAB; 2% for MCEE; and 1% for MMADHC. Due to the genetic heterogeneity of MMA, there are no specific mutation hotspots although specific mutations among various populations have been observed.2,3 The human genome mutation database, HGMD professional 2014.4, currently lists a total of 248 mutations in the MUT gene.8 Approximately 70% of the mutations detected in this gene are missense or nonsense mutations. PolyPhen and SIFT predicted that both mutations identified to be potentially pathogenic, and for this reason, it is highly likely that both mutations are responsible for the MMA phenotype presented in both patients. In patient 1, the c.329A>G;p.Y110C mutation is located in the alpha/beta chain catalytic domain of the protein, whereas in patient 2, the (c.2200C>T;p.Q734X) is located in the cobalamin binding domain of the protein. This mutation is nonsense and leads to a premature stop-codon. In contrast, most but not all mutations reside in the cobalamin binding domain of the mutated protein were associated with residual enzyme activity.9 There are 2 recognized enzymatic subtypes of MMA caused by MUT gene mutations, mut0 with a complete absence of the enzyme activity and mut- with residual enzyme activity.1 We cannot make a strong statement regarding the enzymatic subtype of our patients and their B12 responsiveness as the relevant cellular and biochemical studies required to characterize them precisely were not carried out. These studies include in vivo and in vitro vitamin B12 responsiveness, cobalamin distribution assays, 14C propionate incorporation assays, and complementation analysis.1,2
Compared to most patients with MMA from different ethnicities who are compound heterozygous, our 2 patients were homozygous for MUT gene mutation.1,4 This is not surprising as both parents are first cousins. Also, the prevalence of consanguinity in Saudi population reaches 56% that predisposes to the expression of homozygous mutant genes.10 Similarly, homozygous mutations in MUT gene were previously reported in both mut0 and mut- patients.3,4 The predominant compound heterozygous genotype makes genotype/phenotype correlation difficult as some patients who carry 2 mutations may have one mut- while the other is mut0. This leads to unpredicted enzyme level and possibly diverse clinical phenotype.2
In conclusion, we report 2 novel mutations in the MUT gene causing isolated MMA in 2 Saudi infants.
Clinical Practice Guidelines
Clinical Practice Guidelines must include a short abstract. There should be an Introduction section addressing the objective in producing the guideline, what the guideline is about and who will benefit from the guideline. It should describe the population, conditions, health care setting and clinical management/diagnostic test. Authors should adequately describe the methods used to collect and analyze evidence, recommendations and validation. If it is adapted, authors should include the source, how, and why it is adapted? The guidelines should include not more than 50 references, 2-4 illustrations/tables, and an algorithm.
Footnotes
Disclosure. This study was supported by the College of Medicine Research Center, Deanship of Scientific Research, King Saud University, Riyadh, Kingdom of Saudi Arabia.
- Received April 23, 2015.
- Accepted June 30, 2015.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.