


Abstract
Objectives: To identify genetic defects in an Omani family diagnosed with deafness.
Methods: A cross-sectional association study was conducted at the Department of Biochemistry, College of Medicine and Health Sciences, Sultan Qaboos University, Al-Khoud, Oman and the Centre of Medical Genetics, University of Antwerp, Antwerp, Belgium between August 2010 and September 2014. Microsatellites markers for nine non-syndromic genes were used to genotype the defective locus using the extracted DNA from family members. Sanger sequencing method was used to identify the disease causative mutation. Eazy linkage 5.05 was used to calculate the logarithm of odds score. Lasergene suite was used to detect the mutation position, and Phyre2, SMART, Rasmol, and GOR IV were used to predict the effects of the defect on protein structure and function.
Results: The disease was linked to markers located on chromosome-2 and covering the OTOF (DFNB9) gene. A novel missense mutation that changed nucleotide C to G at position c.1469 and consequently the amino acid Proline to Arginine (P490R) on exon 15 was detected. Protein modeling analysis revealed the impact of the mutation on protein structure and the relevant C2C domain. The mutation seems to create a new protein isoform homologous to the complement component C1q.
Conclusion: These findings suggest that the mutation found in C2C domain of the OTOF gene is likely to cause deafness in the studied family reflecting the importance of C2 domains of otoferlin in hearing loss.
Hereditary hearing loss is a heterogeneous group of a complex disorder with an overall incidence of one in every 500 newborns presented as syndromic and nonsyndromic forms.1 The nonsyndromic pre-lingual deafness is mainly inherited as an autosomal recessive trait in more than 80% of patients,2 60% of which have a family history related to hearing loss, or a confirmed genetic etiology.3 The high rate of consanguineous marriages in the Arabian Peninsula countries is a crucial factor in promoting a spectrum of genetic disorders including nonsyndromic deafness. In the Sultanate of Oman, where the rate of consanguinity is estimated to be 55%,4 a national retrospective analysis from 1986 to 2000 revealed that 70% of the deafness cases have parents of consanguineous marriages.5 During the last decade, molecular genetics has achieved advanced understanding of mechanisms ruling the setup of deafness and identified a number of associated genes loci. For nonsyndromic hearing loss, approximately 170 loci representing approximately 75 genes have been mapped so far.6 Based on their functions, these genes were categorized into several groups including cochlear ion homeostasis, hair bundle morphogenesis, adhesion, scaffolding, exocytosis, signaling at auditory ribbon synapse and transcription.1 The OTOF gene (DFNB9; MIM601071), a member of ferlin family, located on the short arm of chromosome 2 at position 2p23.1 and encoding for the otoferlin protein, is considered one of the relevant genes associated with non-syndromic deafness.7 In human brain and ear cochlea, 2 otoferlin isoforms were identified. A long isoform of 227 kDa encoded by 48 exons and containing 6 C2 domains (C2A-C2F) of 1997 amino acids, and a short isoform of 149 kDa containing 3 C2 domains (C2D-C2F) of 1307 amino acids.8 The C2 domain is one of first 4 domains (C1-C4) to be reported on mammalian protein kinase C (PKC), a Ca2+-dependent protein of approximately 130-140 amino acids long,9,10 All C2 domains share a common structure formed by a duplicated 4-stranded antiparallel ß-sheets connected by variable loops.11 Calcium binds either 2 or 3-binding sites, characterized by a cluster of the negatively charged aspartic acid residues, located between the loops at one end of the domain.12 Calcium binding plays 2 major roles in proteins activity. It either induces an intra/inter domain conformational changes, or, it bridges the C2 domain with different ligands and substrates including phospholipids, inositol polyphosphates, and intracellular proteins involved in signaling pathways.11,13 Otoferlin was found to be highly expressed within inner-hair-cells sensory receptors of the auditory system detected in the vestibular system and the outer hair cells of the cochlea.14-16 In 1999, the first OTOF nonsense homozygous T-A mutation was identified on exon-18 in 4 unrelated Lebanese families affected with a pre-lingual severe to profound form of sensorineural deafness.7 Since then, a plethora of mutations (>90 cases), all associated with deafness, were reported.17 Interestingly, studies on different otoferlin C2 domains including C2C revealed that a single amino acid change and protein truncation on C2 domains were strongly associated with profound deafness.8,18,19 Although the exact mechanism(s) ruling the otoferlin function is still under debate, it is thought that in the presence of calcium, the C2 domain plays a significant role in the release of hair-cell neurotransmitters between cochlear inner-hair-cells (IHCs) and the auditory nerve.20 The exocytosis of hair-cell vesicles and the neurotransmitters releasing process requires an interaction between a group of plasma membranes SNARE proteins like synapbtobrevin 2, syntaxin 1A and 1B, SNAP-25 and the otoferlin C2 domains.21 In this study, we report the screening of an Omani family affected with nonsyndromic deafness for putative genes linked to the disease. We analyzed 9 well-known deafness loci using genetic linkage analysis. The OTOF gene emerged as a potential candidate responsible for deafness in this family and was selected for genescan analysis followed by sequencing the complete coding area composed of 48 exons. Molecular modeling was used to predict the effects of the mutation in the OTOF gene on protein structure and function.
Methods
This is a cross-sectional association study conducted at the College of Medicine and Health Sciences, Sultan Qaboos University, Muscat, Oman. and the Centre of Medical Genetics, University of Antwerp, Antwerp, Belgium between August 2010 and September 2014. It was approved by the College of Medicine and Health Sciences Research and Ethics Committee, Sultan Qaboos University, Muscat, Oman.
An Omani family composed of 10 members, mother, father, 3 female, and 5 male siblings were diagnosed with sensorineural, non-syndromic hearing impairment at the Department of Ear, Nose, and Throat (ENT), Al Nahdha Hospital, Muscat, Oman. The degree of hearing loss in the family was on the scale of >61-80 dB hearing loss as determined by standard pure tone audiometry (PTA). We classified them as severe to profound sensorineural hearing loss (SNHL). Tympanometry, middle ear muscle reflex (MEMR) showed bilateral type A with ipsilateral stapedial reflex absent and auditory brainstem response (ABR) audiometry showed profound hearing loss. The family was subjected to linkage screening for 9 known deafness loci: DFNB1 (GJB2), DFNB2 (MYO7A), DFNB3 (MYO15), DFNB4 (SLC26A4), DFNB7 (TMC1), DFNB8 (TMPRSS3), DFNB9 (OTOF), DFNB12 (CDH23), and DFNB21 (TECTA). We used Qiagen kit (Qiagen, Hilden, Germany) for DNA extraction from peripheral blood samples from the 10 members of the affected family as well as 100 healthy individuals taken as controls. The 9 deafness loci investigated were genotyped using microsatellite markers. Linkage analyses were carried out using the Eazy Linkage 5.05 program package (http://en.freedownloadmanager.org/Windows-PC/easyLINKAGE-Plus-FREE.html) and the 2-point logarithm of the odds ratio (LOD score) were calculated using super link v 1.6 program. The LOD score was higher than 3 for DFNB9 (OTOF) region; therefore, the purified polymerase chain reaction (PCR) products obtained from the 48 OTOF coding exons (GenBank AF183185.1) were sequenced using ABI PRISM Big-Dye terminator cycle sequencing premix kit (PE Applied Biosystems, Austin, Texas, USA). The analysis of sequences was performed using sequence analysis software Lasergene suite (DNASTAR, Inc, Madison, Wisconsin, USA) package. To validate the obtained sequence data, DNA from the 100 unrelated normal hearing control individuals was sequenced and compared with patients’ sequence data. Amplification of all DNA samples was performed using PCR and specific primers listed in Table 1.
Primers for exon 15 of otoferlin.
In order to predict the impact of amino acid changes resulting from the missense variant mutation on the 3 dimensional structure of otoferlin protein and its biological function, Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index), SMART (http://smart.embl-heidelberg.de/), Rasmol (http://www.openrasmol.org/), and GOR IV (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_gor4.html) computer protein analysis programs were used.
Results
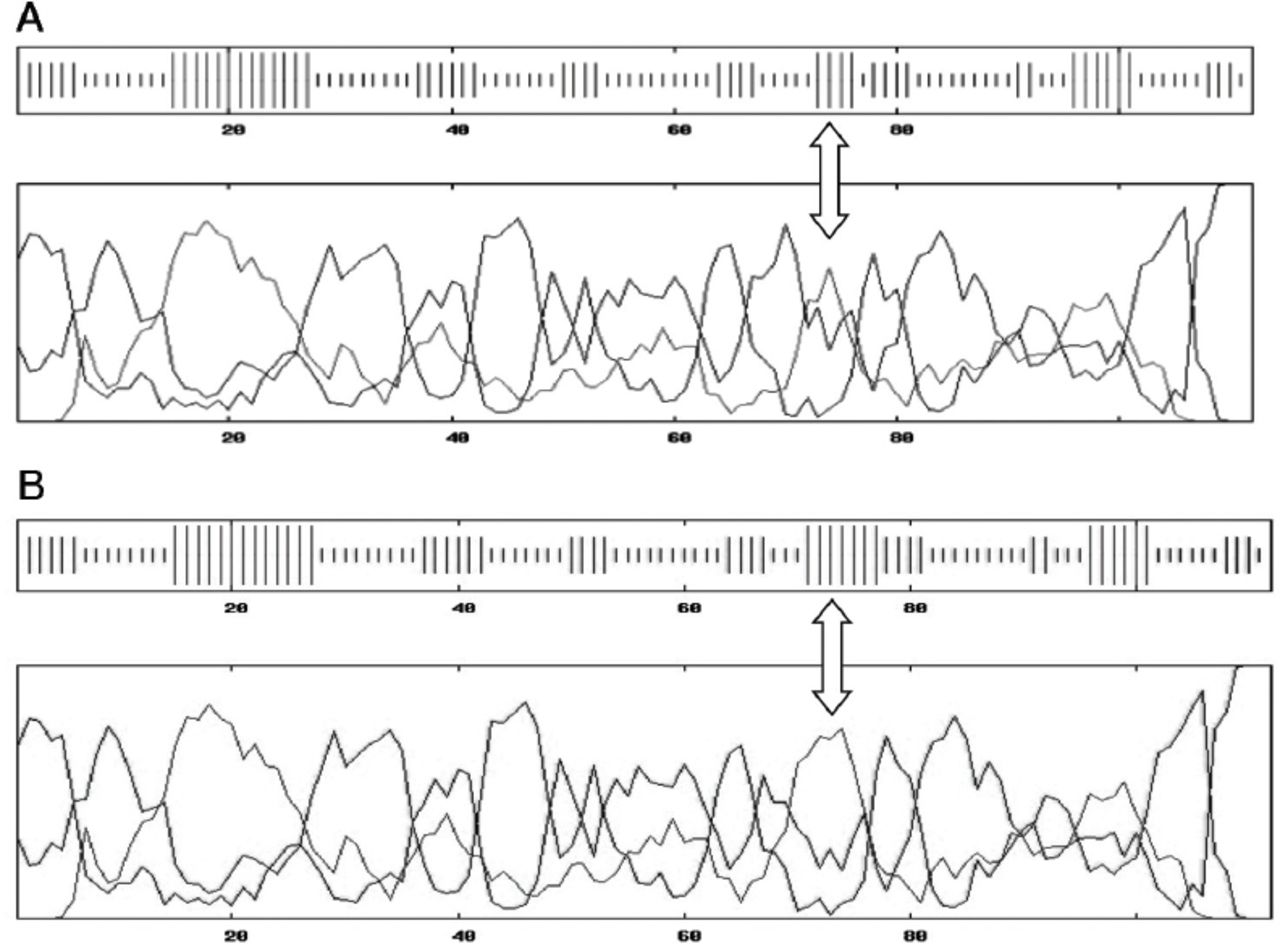
Based on the data obtained from the 10 members of the affected family, a pedigree was constructed displaying the positions of the microsatellite linkage markers D2S2144, D2S2223, D2S365, and D2S174 used to identify the OTOF gene on chromosome 2 (Figure 1). A missense mutation was located at nucleotide position c. C1469G on exon 15 causing a change of the amino acid proline-490 to arginine (P490R). While the parents were carriers of the disease, the affected children were all homozygous for the P490R mutation (Figure 2).

Co-segregation of microsatellite alleles for DFNB9 (OTOF) in the Omani family affected with DFNB9. Microsatellites DNA markers D2S2144, D2S2223, D2S174, D2S365, and D2S366 (the area of OTOF gene) were segregated with the family. Individuals 1-2: heterozygous parents, 3-7: homozygous affected, and 8-10: normal family members.

Electropherogram showing C.1469 substitution mutation C<G in the affected (homo-affected), CC (homo-normal) and CG (heterozygous parents).
Interestingly, none of the unrelated Omani controls with normal hearing were found to harbor the mutation. In order to assess the effect of the P490R mutation on the structure and function of otoferlin protein, we used SMART genetics software to predict the putative changes in the protein by replacing the amino acid proline with arginine. Structural analysis revealed that 6 C2 domains (CA-CF) formed the backbone structure of otoferlin. The C2C seemed to be the most affected domain (Figure 3). In wild type otoferlin, the C2C domain displayed phosphatidylinositol 3-kinases (PI3K-C2) like isoform structure (Figure 3A), whereas the mutated form of C2C domain indicated the presence of the complement C1Q-like structure (Figure 3B).

A diagram showing the A) 6 C2 domains of otoferlin (CA-CF) and B) the amino acids positions of the domains in the protein. 3) Wild type C2C domain contains PI3K-C2 while D) the mutated type has an extra isoform CIQ.
Comparison between the secondary structure of domains in the wild type and the mutated form using GOR IV software revealed a noticeable difference. The replacement of proline with arginine seemed to result in the reduction of coils-beta turns (from 54.5% in the wild type to 51.8% in the mutated form), and in an increase in alpha helix (from 20.5% in the wild type to 23.21% in the mutated form) of C2C domain of otoferlin (Figures 4A and 4B). This is interesting as proline is found in the beta turns (coils) of protein structure. Protein homology database file (PDB) was created for both forms, the wild type as well as the affected forms using Phyre2 and Rasmol software. The 93 residues of the C2C domain of wild type and the mutated forms were compared with extended synaptotagmin-2. C2a and C2b domains of synaptotagmin-2 were used as a reference. While the wild type domain showed approximately 88% of homology, the homology in the mutated domain dropped to 83% with a confidence of 99.7%.

Otoferlin C2C wild type secondary structure. The arrow shows the presence of coils-beta turns and alpha helix. Otoferlin C2C mutated type secondary structure. The arrow shows a reduction of coils-beta turns and an increase in number of alpha helix. lllla-helix, llll Extended strand (ß-strand), llll Other states (Coils – Beta turns)
Furthermore, protein crystallization analysis showed more cohesiveness in the wild type domain when compared to the mutated form (Figures 5A and 5B). These data suggest that the secondary structure of the C2C domain, considered as the backbone of otoferlin, was altered as a consequence of proline to arginine substitution.

Ribbon and space-filling models of wild-type and mutated C2C domains of otoferlin. A) The wild type is more cohesive than B) the mutated form.
Discussion
We investigated an Omani family affected with deafness using genetic linkage analysis to identify putative genes causing the disease. The obtained data revealed a linkage to OTOF gene located on chromosome 2. The 2 points linkage parametric analysis LOD score was significantly higher than 2.75, which confirmed true disease linkage. Sequencing of the 48 exons composing the OTOF gene in the 10 members of the affected family identified a missense mutation changing the nucleotide “C to G” at position c.1469 on exon 15. This mutation resulted in the substitution of the amino acid proline to arginine (P490R). Proline is very rigid, neutral in charge with a hydrophobicity index of -1.6, and has a potential to induce an appropriate backbone conformation required for the protein optimal function. Proline is often localized at the end of the alpha helix, between beta sheet turns and the main protein loops. This provides a tight structure that stabilizes ß-sheet orientations and thus anchors the N-terminus to the main body of the protein.22 These features enhance protein-protein or protein-phospholipid interactions and mediate signal transduction. It has been previously reported that the presence of one or more hydrophobic amino acids in the loop structure, stimulates domain calcium binding, reflecting the importance of domain electrostatic potential changes in accelerating protein membrane interaction.23 Arginine, on the other hand, is a positively charged amino acid, bigger in size than proline, and with a lower hydrophobicity index (-4.5). Such properties would certainly shape the conformational structure of otoferlin domain differently than proline does.24 To estimate the impact of proline to arginine amino acid change on otoferlin structure, modeling analysis using available software such as GOR IV, SMART, Phyre2 and Rasmol were performed to reveal that while the beta turns have reduced, the alpha helix has become bigger in the mutated form, generating a loose and less adherent loop that is unable to fix the termini to the core protein.
We localized the P490R mutation on the C2C domain, one of the 6 (C2A-C2F) protein domains considered relevant for the stereochemistry of otoferlin. Interestingly, a mutation at the same position was reported by Mirghomizadeh et al25 on a Turkish consanguineous family where proline was substituted with glutamine instead (P490Q) and similar findings were observed. Using SMART software, we identified that the mutated area is similar in structure to phosphatidylinositol 3-kinase (PI3K-C2). The PI3K phosphorylates phosphatidylinositol 3 phosphate (PI3P), a principal component of PI signaling pathway, important in mediating membrane fusion, trafficking and cell granules exocytosis.26-28 Moreover, PI3K has also been reported to stimulate Kv7.1-KCNE1in Xenopus oocyte.29 Kv7.1 encodes a voltage-gated potassium channel, which enables a potassium current after electrical depolarization of the cell membrane. Interestingly, such channels contribute to the release of potassium to the endolymph in the inner ear.30 This would balance the “in/out” inner ear endolymph potassium concentration. Kv7.1gene knocked out mice displayed a non-syndromic deafness phenotype and severe anatomic disruption of the cochlear and vestibular end organ.31 These data suggest that Kv7.1 is likely an important player with a crucial role in the development of the inner ear. Moreover, PI3K is thought to promote the trafficking of voltage-dependent calcium channels of plasma membrane during the event of exocytosis,32 suggesting that C2C-PI3K-C2 like domain has an important role in the inner ear exocytosis and development. Despite the efforts to study the exact function of C2 domains, the C2C domain molecular interactions with neurotransmitter secretion remains unresolved. The closest structural and biochemical similarity to otoferlin that has been studied thoroughly is synaptotagmin-1. Synaptotagmin contains 2 domains C2a and C2b where C2a is highly expressed in the neural tissues and regulates the fusion step of synaptic vesicle exocytosis. The 2 synaptotagmin domains are highly interactive with phospholipids PIP2 and PIP3 during the synaptic vesicles fusion step.33 Otoferlin, was predicted to operate through a similar mechanism of interaction.34
Otoferlin as IHC transmembrane protein plays a key role in mediating the presynaptic membrane components interaction. Calcium and intracellular lipids are the main 2 elements to optimize the main function of otoferlin. The role of intracellular calcium-C2 domain binding activity is not clearly understood despite several studies conducted to hypothesize the importance of calcium in the mediation of neuro vesicle-membrane fusion. The process of IHC neurotransmitter release requires interaction of several presynaptic membrane contents. Studies revealed that C2A domain of otoferlin has no calcium binding property whereas the other otoferlin domain vary in their binding affinity to calcium.20,21,35 Only C2C and C2F have binding affinity with the phospholipid phosphatidylinositol 4,5 bisphosphate (PIP2), which is calcium independent.36 However, influx of calcium increases the binding affinity of the domains to PIP2 more than 10 fold and conversely, binding of PIP2 increases the affinity of C2B of synaptotagmin-1 to calcium more than 40 fold.37 The binding property of the 2 otoferlin domains with acid phospholipids may indicate how crucial the interaction of C2C domain is in promoting neurotransmitter release as well as it may reflect the independence of domain function. This hypothesis is supported by the finding that when 2 lysines (K478 and K480) of the conserved C2C polybasic region were mutated to alanine, this resulted in weakened binding with PIP2.36 Excess of intracellular calcium concentration motivates the binging of otoferlin domains with the presynaptic membrane SNARE motif sites suggesting the importance of calcium in mediating the process of vesicle fusion.
This study is in agreement with the previous study25 on hearing loss of the Turkish family. A structural comparison between P490R and P490Q was also conducted and no differences were detected (results not shown). The otoferlin C2C domain might be a crucial region for otoferlin optimum function and any aberration affecting this domain could be responsible for non-syndromic hearing loss seen in the case of the studied Omani family.
Study limitations
This study uses computer modeling analysis to predict the impact of the mutation on otoferlin structure. To confirm this, future protein functional studies will be necessary.
In conclusion, the protein otoferlin has been reported to have an active role in hair cell vesicle exocytosis and the release of synaptic neurotransmitters through C2 domains. Protein modeling analysis revealed the impact of a novel missense mutation that changed nucleotide C to G at position c.1469 and consequently the amino acid Proline to Arginine (P490R) on exon 15 on protein structure and the relevant C2C domain. The mutation seems to create a new protein isoform homologous to the complement component C1q, which suggest that the mutation found in the C2C domain of the OTOFgene is likely to cause of hearing loss detected in the family investigated.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company. This project was supported by the College of Medicine and Health Sciences, Sultan Qaboos University, Muscat, Oman (Grant IG/MED/BIOC/08/01).
- Received March 9, 2016.
- Accepted July 11, 2016.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.