


Abstract
Glycogenic hepatopathy is a rare condition that causes significant hepatomegaly and elevated liver enzyme levels in uncontrolled type 1 diabetic patients. It develops due to excessive accumulation of glycogen in the hepatocytes. It is typically reversible with good glycemic control and rarely progresses to mild fibrosis, but not cirrhosis.
Hepatomegaly with or without elevated transaminases levels is a common condition in patients with uncontrolled type 1 diabetes mellitus (DM). Glycogenic hepatopathy (GH) is a rare disease that develops due to excessive accumulation of glycogen in the hepatocytes, leading to hepatomegaly and elevated transaminases levels in patients with uncontrolled type 1 DM.1 Glycogenic hepatopathy is a benign and reversible condition with good prognosis, in contrast to non-alcoholic fatty liver disease (NAFLD), which is more common in type 2 DM and may lead to liver fibrosis. Although GH is well-described, it is still underdiagnosed. In this report, we describe the clinical, biochemical, and histopathological features in a Saudi girl with GH.
Case Report
A 6-year-old girl with a 4-year history of type 1 DM on insulin therapy (Insulin glargine [Lantus®] 7 units at bedtime and Insulin aspart [NovoRapid®] 5 units premeal), she presented with hepatomegaly and elevated liver enzymes since 6 months. Her glycemic control was suboptimal, with persistently elevated glycated hemoglobin A1c (HbA1c level 11%) for the prior 2 years (normal range [NR]: 3.9-6.7%). There was no history of jaundice, itching, change in urine or stool color, fever, rash, bowel complaint, contact with sick patients, exposure to animals, travel, or use of drugs/herbals. Her past history was unremarkable, with normal developmental history. The family history was unremarkable for liver disease. Physical examination showed normal growth parameters (weight 17 kg (10% percentile, height 109 cm (5th-10th percentile). She had hepatomegaly (liver span 14 cm), but no stigmata of chronic liver disease. Examination of other systems was unremarkable. The initial test results were as follows: alanine aminotransferase (ALT), 205 (NR 20-65 units/L), aspartate aminotransferase, (AST) 367 (NR 10-31 units/L), gamma-glutamyl transferase (GGT), 500 (NR 5-55 units/L), and alkaline phosphatase (ALP), 221 (NR 175-476 units/L). Liver synthetic function (bilirubin, albumin, and coagulation profile) and lipid profile were normal. Work up for infectious causes (hepatitis B virus, hepatitis C virus, cytomegalovirus, and Epstein-Barr virus) were negative. Thyroid stimulating hormone (TSH), celiac screen, ammonia, lactate, and metabolic screenings were all normal. In addition, investigations for autoimmune hepatitis and Wilson’s disease were unremarkable. Ultrasound of the abdomen showed hepatomegaly (17.9 cm) with normal echogenicity and no focal lesions; the spleen and kidneys were unremarkable.
With strict glycemic control, the patient’s liver enzymes returned to normal levels, without further intervention. Four month later, she presented with diabetic ketoacidosis (DKA) and acute hepatitis (ALT: 1,408 units/L, AST: 856 units/L, GGT: 1,007 units/L, ALP: 408 units/L), but normal liver synthetic functions. Biopsy showed preserved liver architecture, but the hepatocytes exhibited marked cytoplasmic swelling and prominent plasma membranes. No inflammation or fibrosis was found. Sinusoidal compression by the swollen hepatocytes lent a “paved” appearance to the liver parenchyma. Additionally, numerous hepatocytes exhibited glycogenized nuclei and abundant periodic acid-Schiff (PAS) stain-positive cytoplasmic glycogen deposits, which were not visible after digestion with diastase to confirm glycogen deposition (Figure 1A-C). Genetic panel testing for glycogen storage diseases known mutations was negative. Ultimately, a diagnosis of GH was made. With good glycemic control, both liver size and liver enzymes were significantly reduced; however, the transaminases levels kept fluctuating according to the glycemic control as they increase to the level of hundreds during the periods of poor glycemic control (as indicated by HBA1C level) and then regress to normal/near normal levels with good glycemic control.
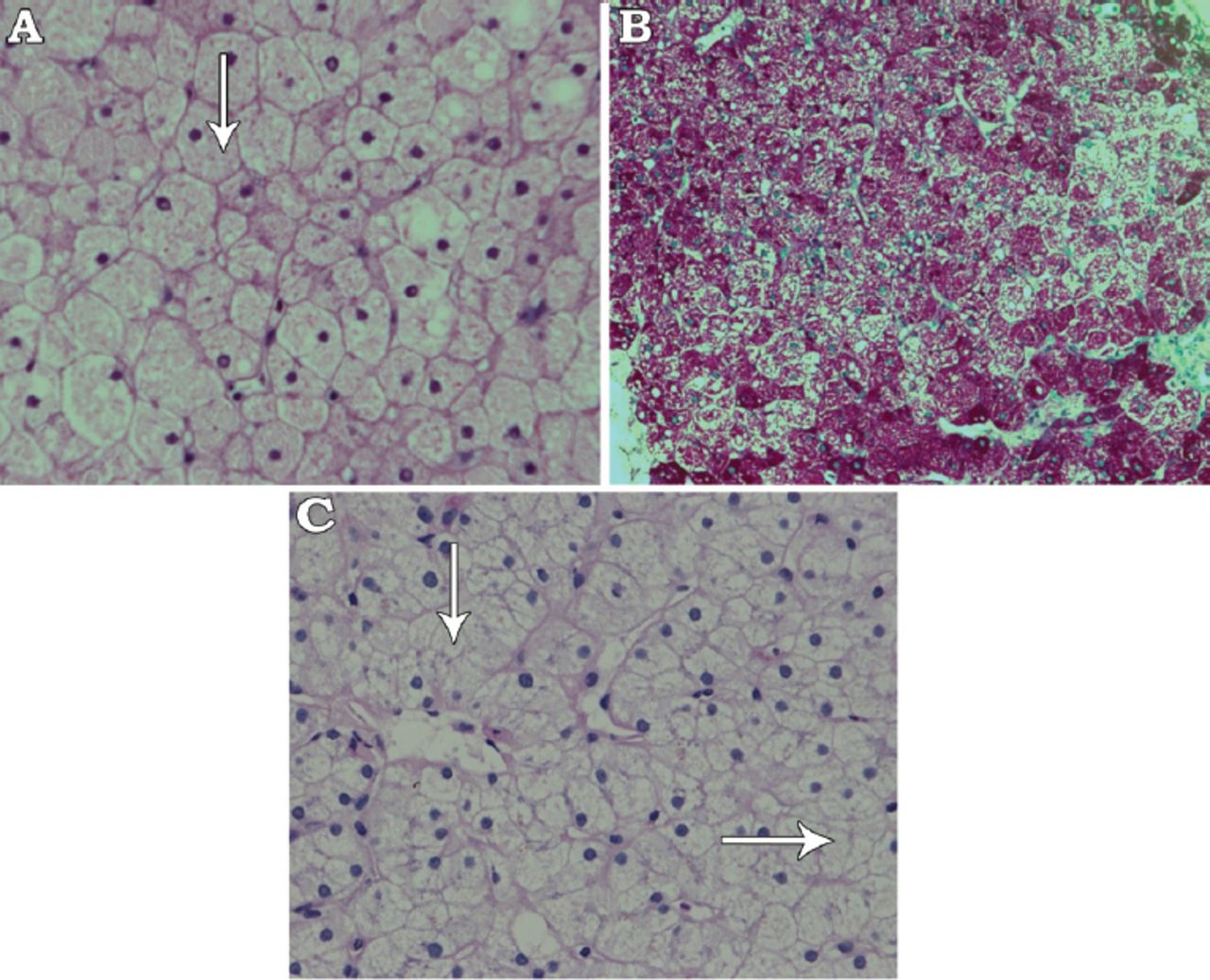
Histopathological section of the liver showing: A) Hepatocytes with clear and fine granular cytoplasm (white arrow) (hematoxylin and eosin stain ×400), B) Periodic acid-Schiff (PAS) stain showing large amount of dark red-colored glycogen in hepatocytes (PAS stain ×200), and C) Complete digestion of glycogen after adding diastase stain and reappearance of clear hepatocytes (white arrow) (PAS stain and diastase ×400).
Discussion
Hepatomegaly with or without raised transaminase levels is common in patients with DM. Glycogen-induced hepatomegaly in type 1 DM was described first by Mauriac in 1930, as a component of a syndrome characterized by hepatomegaly, growth retardation, cushingoid appearance, and delayed puberty. Mauriac syndrome has become rare with the advent of better insulin regimens and improved multidisciplinary care.2 However, there have recently been increasing numbers of case reports describing this condition in both children and adults with type 1 diabetes, but without other components of Mauriac syndrome.1,3 There have also been some cases reported in patients with type 2 DM1.4 The term glycogenic hepatopathy has been proposed by Torbenson et al1 to represent a pattern of glycogen accumulation within hepatocytes, which is characterized by elevated serum transaminases and/or hepatomegaly, in patients with uncontrolled DM. The exact incidence and prevalence of the condition are not well known, as the condition is underrecognized. The clinical symptoms and signs of GH are nonspecific, and include abdominal pain, hepatomegaly, and marked transaminases elevation resembling acute and relapsing hepatitis. Liver synthetic functions are typically preserved.
The hepatic glycogen level is maintained by the balance between glycogenesis and glycogenolysis in the liver. In diabetic patients with poor glycemic control, blood glucose and insulin levels often demonstrate significant fluctuations, with hypo- and hyperglycemic attacks.
In the presence of hyperglycemia, glucose passively diffuses into hepatocytes (insulin-independent mechanism), and is subsequently converted to glucose-6-phosphate, which is then converted to glycogen under the influence of insulin; these changes are exaggerated in the presence of hyperglycemia and over-insulinization, which are believed to represent the primary mechanism in the development of GH.5,6 In addition to GH, there are several differential diagnoses that need to be considered in any patient with DM and hepatomegaly; these include NAFLD, glycogen storage disease (GSD), and autoimmune disorders, such as celiac disease and autoimmune hepatitis, both of which may be associated with DM. While NAFLD is the most likely diagnosis in obese patients with type 2 DM, GH is more common in patients with poorly controlled type 1 DM. It is difficult to distinguish GH from NAFLD neither on the basis of clinical presentation nor by liver ultrasound. So far, liver biopsy is the only diagnostic tool that can differentiate between the 2 conditions.
The clinical presentation of markedly elevated liver enzymes in the background of poorly controlled type 1 DM is highly suggestive of GH. Genetic testing, as performed in our case, can help to rule out GSD. Liver biopsy is the gold standard test for GH diagnosis. The main histological features of GH are marked glycogen accumulation, leading to pale, swollen hepatocytes, with typically minimal inflammation, steatosis, and fibrosis.1 A recent report by Fitzpatrick et al.7 described the histopathology findings in 31 young patients (median age of 15 years) with GH; they reported a high percentage of inflammation (42%) and fibrosis (73%), but these were generally mild. There is still a need for larger-scale and long-term studies to explore the consequences of fibrosis over time. Some case reports described the utility of radiological imaging studies such as computed tomography (CT scan) and magnetic resonance imaging (MRI) in establishing the diagnosis of GH depending primarily on the interval changes in the liver density and its correlation with the clinical picture; however, further larger-scale studies are needed to confirm the findings from these reports, and to establish their sensitivity and specificity for this condition.8,9
Glycogenic hepatopathy is a benign condition that is potentially reversible within 2 to 14 weeks, both clinically and biochemically, with good glycemic control.10,3 The degree of glycemic control required to achieve this result has not been well described, but aggressive insulin therapy is not necessarily required.10,6 Parmar et al,6 reported a case in which an improvement of just 0.6% in HbA1c led to symptomatic relief of abdominal pain and a decline in liver enzymes.
In conclusion, Although it is a well-known complication of poorly controlled type 1 DM, GH is still an underdiagnosed cause of elevated transaminase levels and hepatomegaly in these patients. Physicians should be aware of this condition and should consider it in the differential diagnosis of hepatomegaly with elevated liver enzymes during the workup of type 1 DM patients. Early diagnosis and subsequent implementation of good glycemic control will ultimately lead to an excellent prognosis. Larger-scale and long-term studies are still needed to clarify the incidence and prevalence of the condition, and to tests the utility of noninvasive diagnostic modalities such as MRI in the diagnosis of this condition.
Illustrations, Figures, Photographs
All figures or photographs should be submitted in a high resolution (minimum 300 DPI) electronic version saved in jpeg or tiff format. Original hard copies of all figures may be requested when necessary. Photographs will be accepted at the discretion of the Editorial Board. All lettering, arrows, or other artwork must be done by an artist or draftsman. If arrows are used please ensure they appear in a different color to the background color, preferably black with a white border, or white with a black border. If arrows distinguish different items on the figure then different arrow styles should be used ie. long, short, wide, narrow. Written informed consent for publication must accompany any photograph in which the subject can be identified. Written copyright permission, from the publishers, must accompany any illustration that has been previously published.
Acknowledgment
The authors gratefully thank Dr. Ammar Al Rikabi, Department of Pathology, King Khalid University Hospital, King Saud University, Riyadh, Kingdom of Saudi Arabia for his assistance in providing and reading the histopathological slides.
Footnotes
Disclosure. Authors have no conflict of interest, and the work was not supported or funded by any drug company, This work was supported by the College of Medicine Research Center, Deanship of Scientific Research, King Saud University, Riyadh, Kingdom of Saudi Arabia.
- Received July 24, 2016.
- Accepted October 19, 2016.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.