


Abstract
We are presenting a monozygotic twin brothers presented at different ages with different presentations. Twin-A presented at age of 18 days with salt losing crisis. Investigations revealed high plasma renin with low-normal aldosterone. Adrenocorticotropic hormone stimulation test revealed low 17-OH progesterone at 0 and 60 minutes. Adrenocorticotropic hormone level and serum cortisol were normal, which excluded initial impression of congenital adrenal hyperplasia. He was diagnosed to have isolated primary hypoaldosteronism. At age of 18 months, he was noticed to have hyperpigmentation of lips and gum. Adrenal failure was suspected, and hydrocortisone was added. Twin-B presented at 9 years and 6 months of age with adrenal crisis. Both were having unilateral undescended testis. Adrenal hypoplasia congenita (AHC) was suspected after his twin’s presentation. Molecular analysis for gene study for both of them revealed adrenal insufficiency, NR0B1 (DAX1) gene mutation. In conclusion, gene analysis is important for the diagnosis of AHC and for genetic counseling.
X-linked congenital adrenal hypoplasia (CAH) is a very uncommon genetic disorder that is caused by an abnormality in the DAX-1 gene (dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1 [NR0B1]),1 was first introduced in 1948. Mutations in this gene were proved as the cause of the X-linked congenital adrenal hypoplasia in 1994.5
The DAX-1 gene consists of 2 exons and a single 3.4-kilobase intron. The protein coded by this gene is one of the nuclear receptor family. DAX-1 gene is expressed in many glands including the adrenals, gonads, hypothalamus, and pituitary gland. It controls their development and function. Adrenal hypoplasia congenita (AHC) is a rare disorder inherited as an autosomal recessive or X-linked disease.2 The X-linked AHC is due to DAX-1 gene mutation or deletion. The typical presentation in boys with X-linked AHC during neonatal period or childhood, is primary adrenal insufficiency. Later in life, they may develop different puberty disorders. Classically, delayed or absence puberty due to hypogonadotropic hypogonadism (HHG). Rarely, they may proceed to normal puberty or may develop precocious puberty (peripheral, central, or adrenocorticotropic hormone-dependent).2,3
Case Report
Patient information twin-A
He was born on 13th February 2006, at 37 weeks of gestation to a 39-year-old gravida 6 para 6 mother after uncomplicated twin pregnancy. The parents were second degree cousins of Saudi origins. He has 2 elder brothers and 3 sisters, all are alive and healthy. His birth weight was 2.0 kg, body length was 46 cm, and head circumference was 32 cm. He was discharged with his mother in good condition after 3 days of admission to the nursery for observation.
Clinical findings
At the age of 18 days, he was presented to the emergency department with history of vomiting, poor feeding, and decreased activity with failure to thrive. On physical examination, his weight was 1.66 kg, blood pressure (BP) was 67/51 mmHg, heart rate was 160/min, temperature was 36.2°C, and respiratory rate (RR) was 58/min. He was dehydrated, not dysmorphic with no evidence of hyperpigmentation. Normal systemic examination, with normal male genitalia (Figure 1).
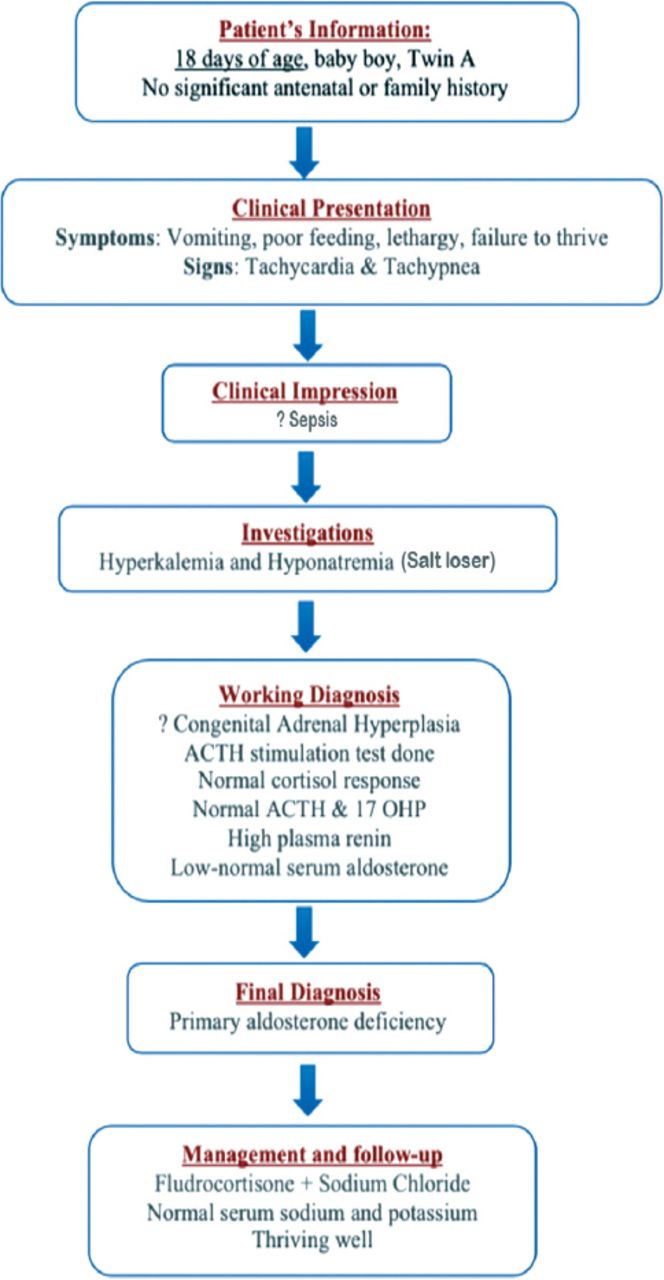
Timeline summarizing the patient’s (Twin A1) information, clinical presentation, clinical impression, investigations, working diagnosis, final diagnosis, and management and follow-up.
Diagnostic assessment
He was admitted to neonatal intensive care unit with impression of sepsis. Laboratory tests showed hyponatremia 128 mmol/L with hyperkalemia 6.2 mmol/L. Septic workup was carried out and were awaited. He was managed with intravenous fluids and antibiotics. He was noticed to have persisted hyponatremia and hyperkalemia, therefore endocrine consultation was requested. A provisional diagnosis of congenital adrenal hyperplasia was made as it is the most common cause of salt wasting at this age group. His endocrinological data revealed unelevated adrenocorticotropic hormone (ACTH), serum aldosterone low-normal while plasma renin was very high. Adrenocorticotropic hormone stimulation test was carried out with 0.25 mg synthectin which showed normal cortisol response at 60 minutes. Seventeen hydroxyprogesterone, dehydroepiandrosterone sulfate (DHEAS), and testosterone were normal. Chromosomal study showed 46 XY (Table 1).
Laboratory findings for twin A at 18 days of age at initial presentation.
Therapeutic intervention
These results ruled out CAH. He was diagnosed to have isolated aldosterone deficiency, and managed with fludrocortisone and sodium chloride orally with excellent response. He was followed up in outpatient clinic regularly and was maintaining normal serum sodium and potassium. He was thriving well (Table 2).
Laboratory findings for twin A at 7 months of age. Adrenocorticotropic hormone stimulation test repeated showed normal cortisol response.
At 18 months of age, he was noticed to have increased pigmentation specially the lips and gum, but was thriving well. Glucocorticoid deficiency was suspected. Urgent ACTH stimulation test was carried out with 0.25 mg synthectin. Basal ACTH >2000 pg/ml. Serum cortisol failed to rise in response to ACTH at 60 min. Seventeen hydroxyprogesterone was normal. Testosterone and DHEAS were normal (Table 3). He was diagnosed to have primary adrenal insufficiency managed with hydrocortisone and fludrocortisone orally. At 18 months of age ultrasound done, showed right testis at the right inguinal area. He underwent orchidopexy for right undescended testis (Figure 2).
Laboratory findings for twin A at 18 months of age. adrenocorticotropic hormonestimulation test.

Timeline summarizing the patient’s (Twin A2) follow-up, diagnosis, management, final diagnosis, and follow up.
Follow-up and outcomes
He was followed up regularly in outpatient department showing normal ACTH and serum electrolytes, and was thriving well. His last visit was at the age of 12 years and 3 months (Table 4). His height was 134 cm (just below 3rd centile), his weight was 29 kilograms (below 10th centile).
Laboratory findings for twin A at 12 years and 3 months.
Patient information twin-B
He is now 12 years and 3 months old boy, his birth weight was 1.8 kg. He was discharged with his mother in good condition. He was growing normally and was not having any significant illness.
Clinical findings
At the age of 9 years and 6 months, his mother brought him to endocrine clinic accompanying his twin-A brother. The mother complained that she noticed him to have progressive weight loss, fatigue, decreased activity, and progressively increasing generalized body pigmentation, which was noticed for 3 months. There was no history of vomiting, abdominal pain, or change in bowel habit. There was no history of preceding infection. On examination, he was alert and conscious. The Glasgow Coma Scale is 15/15, lethargic, dehydrated with generalized marked hyperpigmentation. The body weight was 18 kg (below 3rd centile), the height was 122 cm (below 5th centile). His temperature was 36.5°C, heart rate was 109/min, BP was 104/59 mmHg, RR was 36/min, and oxygen saturation was 100%. Systemic examination was normal except for right undescended testis (Table 5). He was admitted to Pediatric Intensive Care Unit with the impression of adrenal crisis due to adrenal insufficiency. He was managed with intravenous hydrocortisone, intravenous normal saline, kayexalate, and fludrocortisone. Orchidopexy was carried out later (Figure 3).
Laboratory findings for twin B at 9 years and 6 months of age as initial presentation.
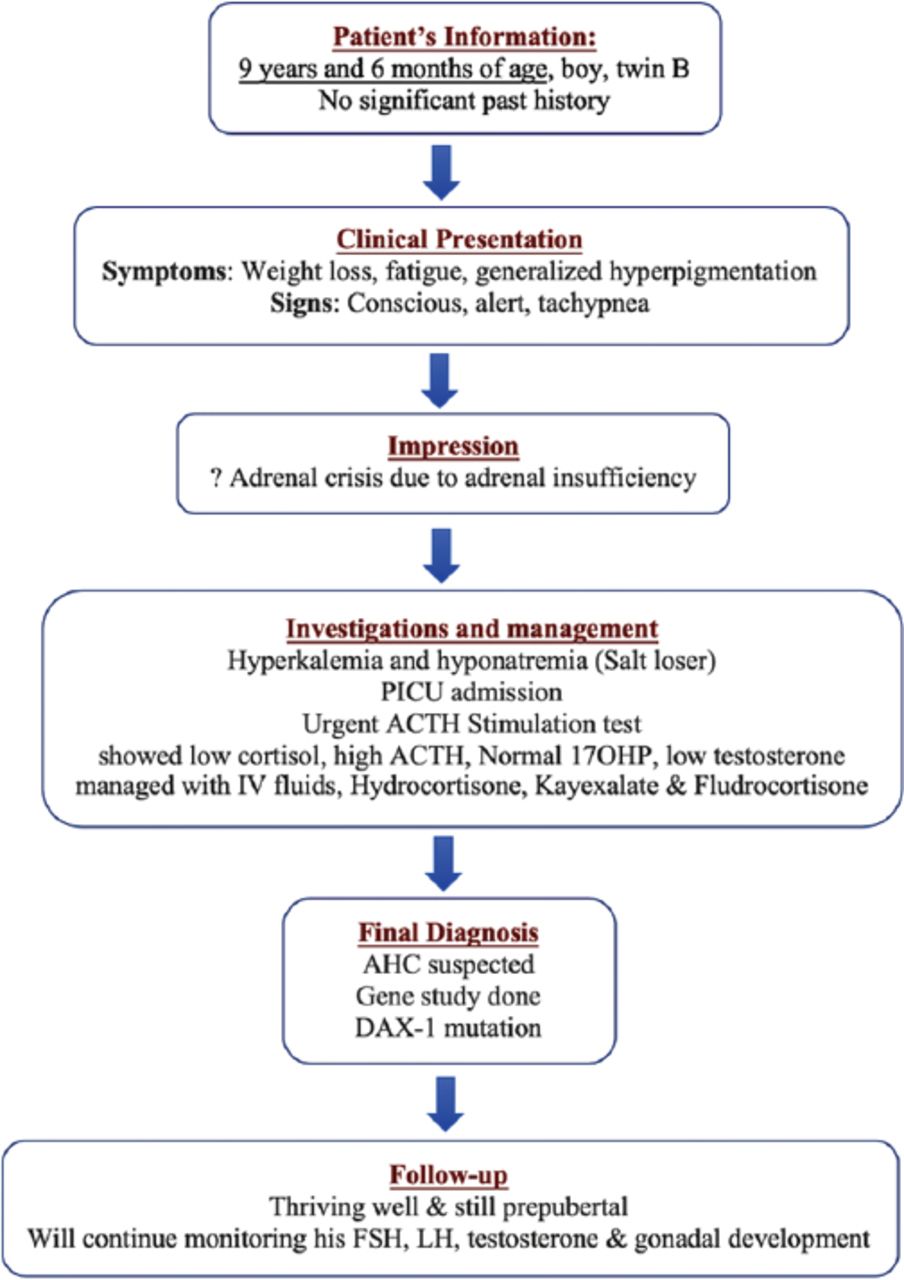
Timeline summarizing the patient’s (Twin B) information, clinical presentation, impression, investigation and management, final diagnosis, and follow-up.

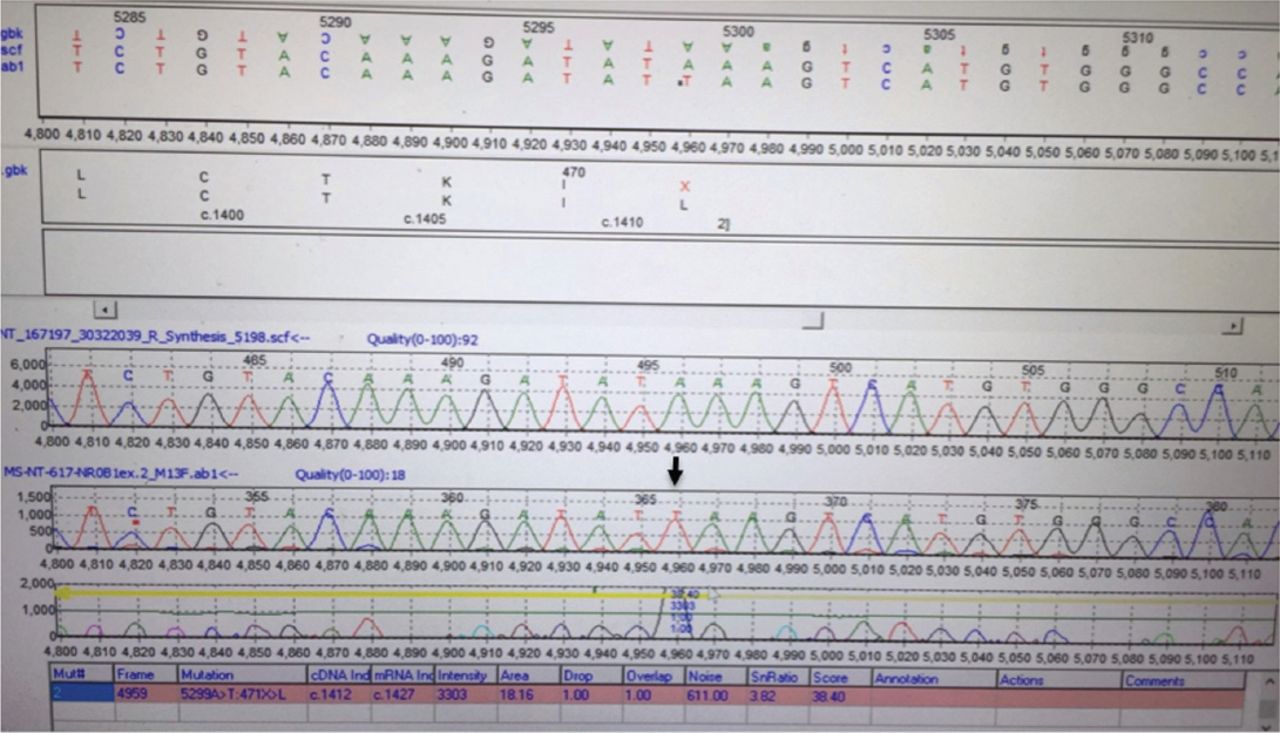
Sequence chromatogram showing a bialleic nonsense mutation changing adenine to thiamine at coding position 1412 (c.A1412T) leading to change of the amino acid Leucine to a stop codon with truncation of the protein at amino acid 471 (p.471 L>X).
Diagnostic assessment
As his twin-A brother was diagnosed to have primary adrenal insufficiency, AHC was suspected and blood samples for gene study were sent for both of them which proved DAX-1 mutation. After 2 months of hydrocortisone replacement the ACTH was 45.37 pg/mL. At 12 years and 3 months of age his wight was 32 kg (above 10th centile), and his height was 135 cm (3rd centile) (Table 6).
Laboratory findings for twin B at 12 years and 3 months.
Therapeutic intervention
Both brothers are now on oral hydrocortisone and fludrocortisone replacement therapy.
Follow-up and outcomes
They are thriving well, repeated hormonal evaluation, particularly serum ACTH was performed and is maintained within the normal reference range. Both of them have normal penile length, and Tanner stage 1 for testis and pubic hair.
Discussion
Primary adrenal insufficiency is a potentially life threatening disorder that can present with salt losing crisis or profound hypoglycemia and requires urgent resuscitation and appropriate steroid replacement. 2 Primary adrenal insufficiency can occur at any age, in the neonatal period, in infancy or in childhood.6 It is difficult to diagnose AHC in a neonate because it is often misdiagnosed as the salt wasting form of congenital adrenal hyperplasia which is the most common etiology for adrenal insufficiency in this age group.2,4 In fact, both of these 2 diseases have different steroid metabolism and can be distinguished from each other by clinical manifestation and genetic features.4
Adrenal hypoplasia congenita is a rare disorder that can be inherited as an X-linked or autosomal recessive pattern.2,4 The exact incidence of AHC is not known, however, for the X-linked form, the incidence is estimated between 1:140,000 and 1:1,200,000 children.2 More than one hundred patients with DAX-1 mutations, have been described.5 DAX-1 mutations are more likely in patients with a positive family history of an affected male.2
X-linked AHC is caused by deletions or mutations in DAX-1 gene (AHC; MIM: 300200),9 the majority of these mutations are frameshift or nonsense mutations leading to truncated DAX-1 protein.2 There is no clear evidence for a genotype-phenotype correlation between a mutation in DAX-1 (NR0B1) and its structural consequence and the clinical phenotype. The age of onset of adrenal failure can vary within the same family, suggesting that other epigenetic factors influence the clinical coarse of AHC1,5 and that’s what we have described in our identical twins. Twin-A presented with salt wasting crisis in the early neonatal period which was followed with glucocorticoid deficiency after 18 months. While twin-B presented with adrenal crisis after many years (at age of 9 years and 6 months) although they were having the same mutation in DAX-1. It has also been observed that in some patients with AHC, the apparent mineralocorticoid deficiency frequently can precede glucocorticoid deficiency7 and this explained why twin-A was initially diagnosed as having isolated aldosterone deficiency. The key treatment during an acute adrenal crisis for these patients are intravenous hydrocortisone as well as normal saline with glucose solutions. These patients require lifelong replacement of glucocorticoids (physiological dose) as well as mineralocorticoids.2
Adrenal hypoplasia congenita is frequently associated with hypogonadotropic hypogonadism (HHG, MIM 1416110), the spectrum of presentation of HHG varies widely from pubertal failure to infertility.2,3,8,9
Mutational (genetic) analysis of DAX-1 is important in any male infant presenting with salt-losing adrenal failure, when steriodogenic disorder (congenital adrenal hyperplasia) and adrenal hemorrhage have been excluded.2 Genetic analysis of DAX-1 gene is very useful for definitive diagnosis of X-linked AHC as well as for genetic counseling in families having DAX-1 mutation with a history of unexplained death of maternal male relatives, highlighting X-linked pattern of transmission.10
We have described an identical twin with DAX-1 mutation who presented at different ages with different presentations. Twin-A presented with isolated mineralocorticoid deficiency during neonatal period, which was followed after 18 months with glucocorticoid deficiency. Furthermore, twin-B was totally normal till the age of 9 years and 6 months when he presented with adrenal crisis. Both of them were having undescended testis requiring orchidopexy and both are still having small testicular size at 12 years of age. They will be followed up closely for their pubertal development as they are likely to have HHG. In conclusion, gene analysis is important for the diagnosis of AHC and for genetic counseling
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received August 6, 2018.
- Accepted November 28, 2018.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.




{kind=link}
{kind=link}
{kind=link}
{kind=link}