


Abstract
Anonychia refers to the absence of nail plates owing to an autosomal dominant or recessive inheritance. Congenital anonychia is a rare condition that may be associated with other ectodermal or mesodermal malformations like epidermolysis bullosa, (deafness, onychodystrophy, osteodystrophy, and mental retardation) syndrome and Iso-Kikuchi syndrome. Here, we report 3 cases with anonychia congenita appearing in different generations of a single family in Kingdom of Saudi Arabia.
Congenital anonychia is a rare condition characterized by the absence of multiple nails or the whole fingernails, toenails, or both, since birth. It may occur as an isolated abnormality or as part of a syndrome or condition. The most common types of anonychia are acquired anonychia and congenital syndromic anonychia, which usually linked to other anomalies compared to the congenital nonsyndromic type.1 The skin in the place of the missing nails is normal and the nail bed and nail matrix or fold are intact in all fingers and toes.2 Complete anonychia generally follows an autosomal recessive pattern and has been allocated to chromosome 20p13 with a mutation in the R-Spondin 4 (RSPO4) gene, and frizzled class receptor 6 (FZD6).3,4
Here, we report 3 cases of complete anonychia congenita running in different generations of a single family which has never been reported in Kingdom of Saudi Arabia (KSA).
This study aims to plan and conduct genetic analysis to detect RSPO4 gene mutations with family history consultation for these patients
Case Report
All of the 3 cases belong to one generation from a single large family of Arabic origin from KSA, with other 4 affected individuals of different generations (Figure 1).
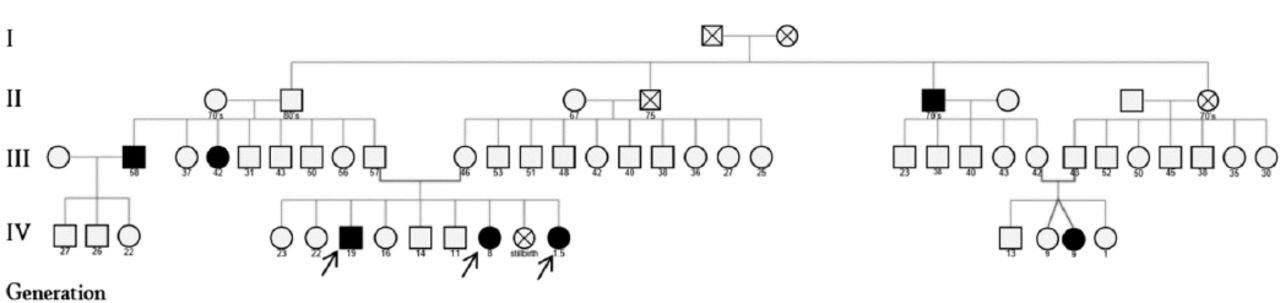
Pedigree of the family with simple congenital anonychia, showing the 7 affected members belonging to different generations. Arrows represent the studied patients.
Patient one
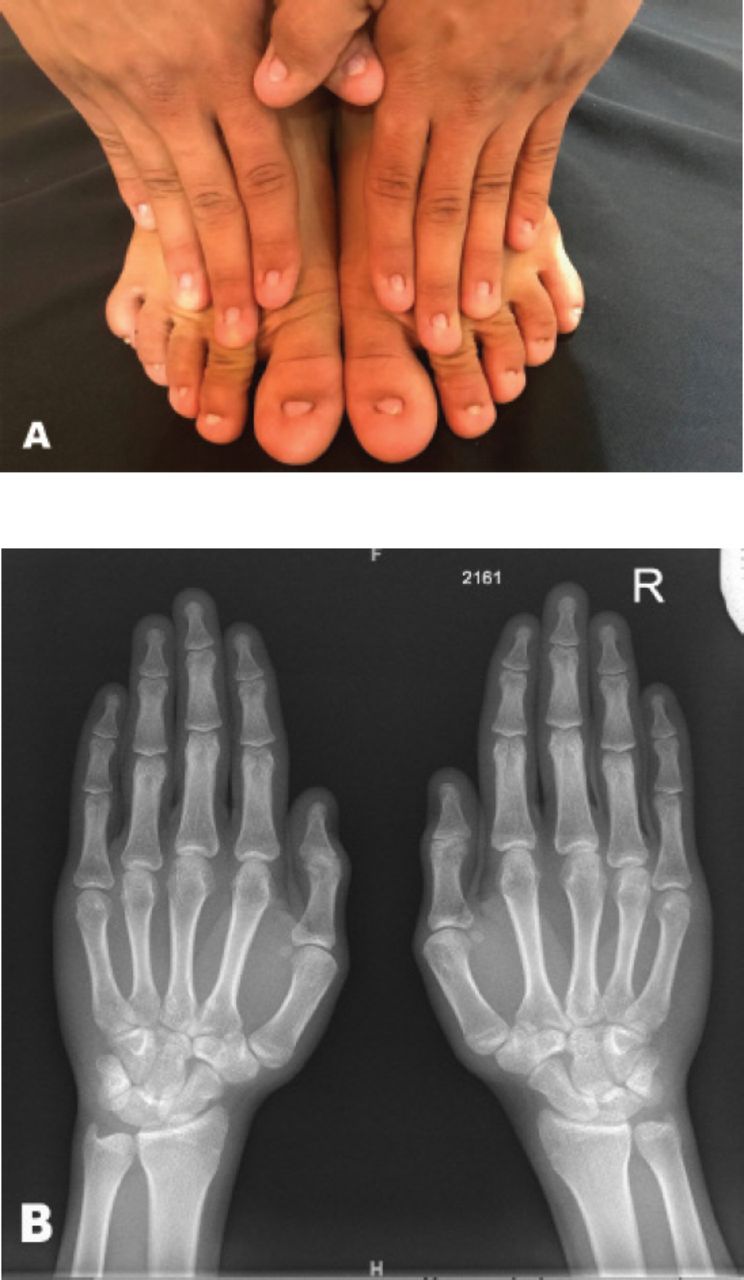
A 19-year-old Saudi male presented with complete absence of all fingernails and toenails since birth. He was born by normal vaginal delivery, full term with normal weight in an uneventful pregnancy. He had normal developmental milestones with no associated physical abnormalities. The parents are first degree relatives and healthy, without any congenital anomalies. No other congenital disorder was known in the family (Figure 2).
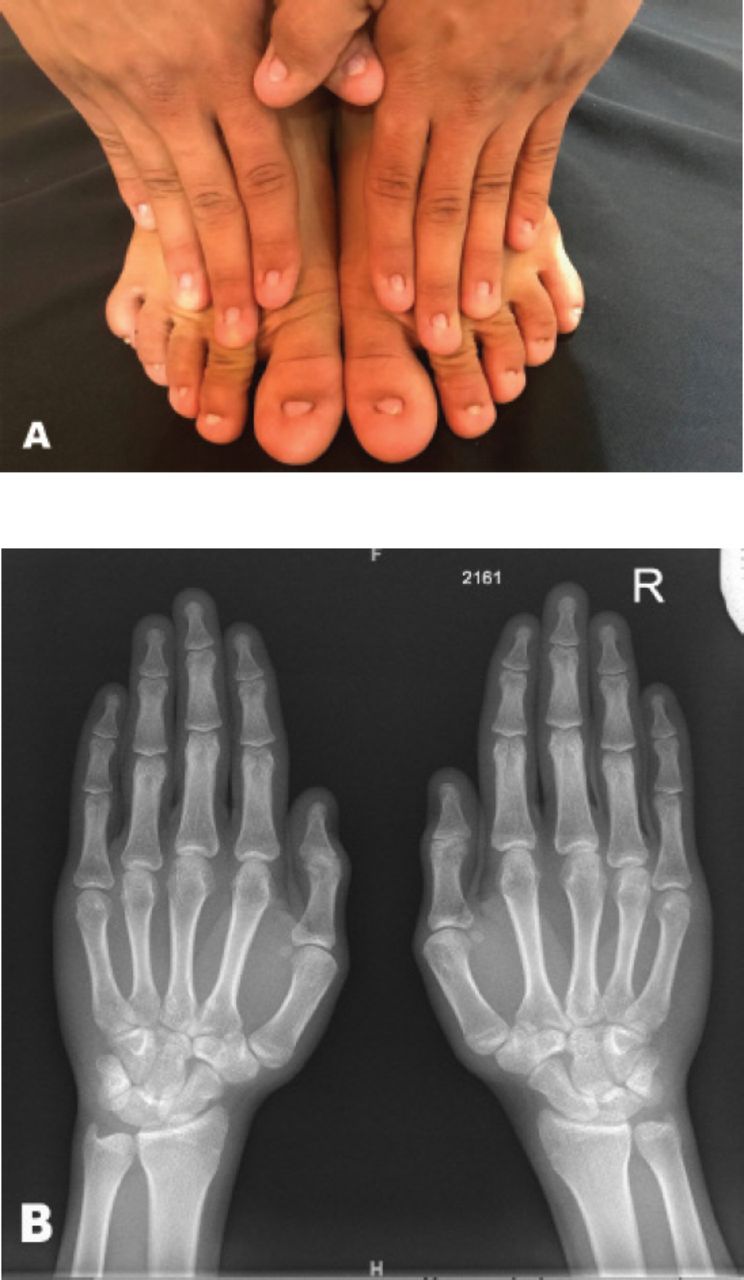
A) Total anonychia of all nails of fingers and toes of patient one. B) Normal bone appearance of both hands as seen in the x-ray report.
Patient 2
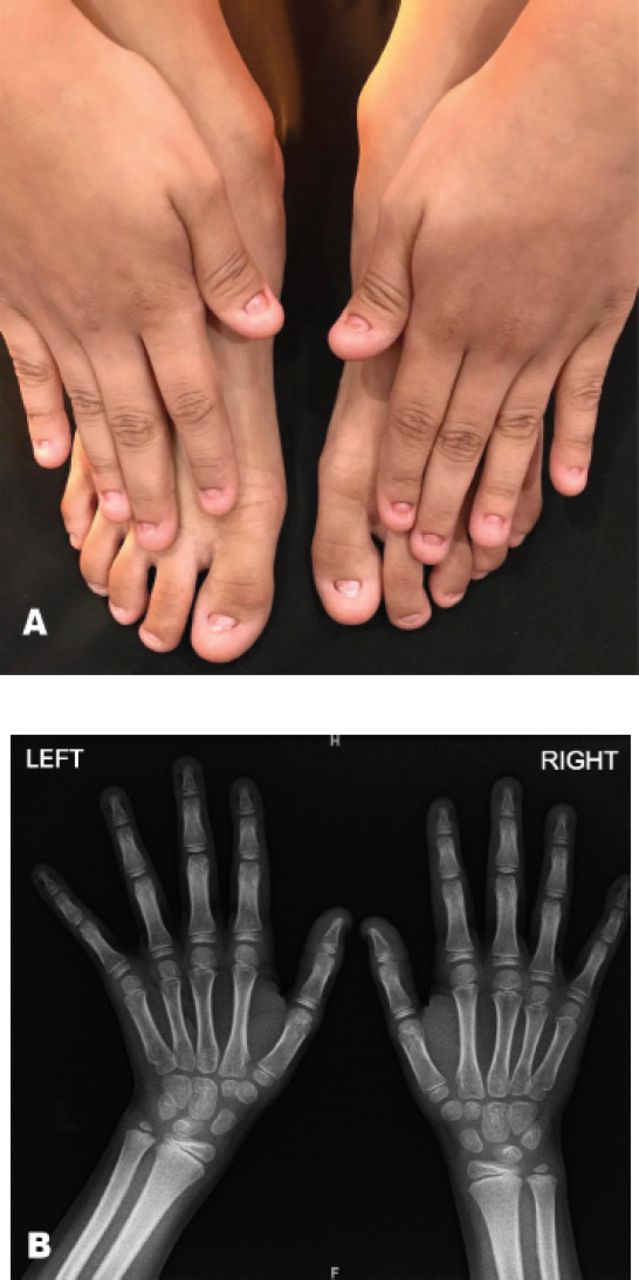
An 8-year-old Saudi female presented with complete absence of all fingernails and toenails since birth. The mother was on 100 mg thyroxine during her pregnancy due to hypothyroidism. The patient had normal developmental milestones with no associated physical abnormalities. The parents are first degree relatives and healthy, without any congenital anomalies. No other congenital disorder was known in the family (Figure 3).

X-ray showing the A) total anonychia of all nails of fingers and toes of patient, and B) Normal bone appearance of both hands.
Patient 3
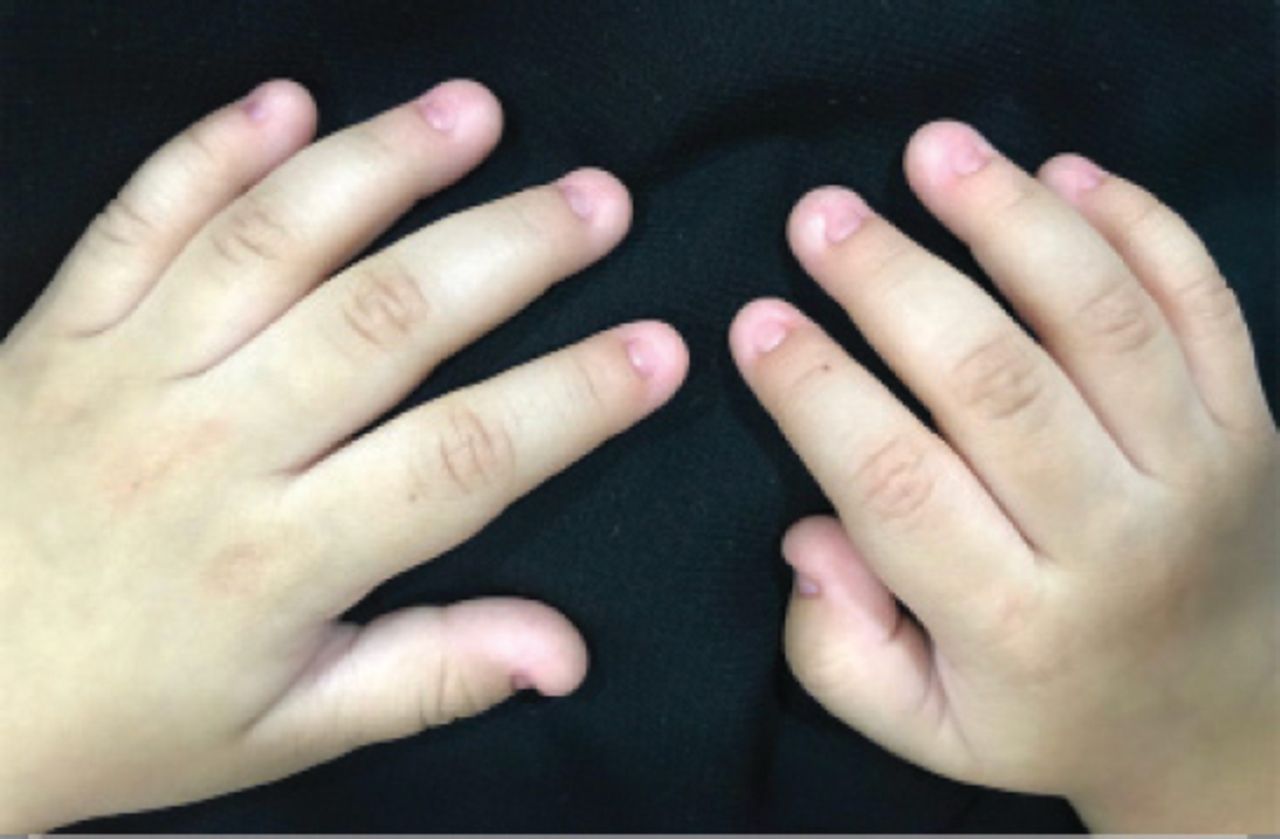
An 18-month-old female infant presented with complete absence of all nails in hands and feet since birth. She was a full-term baby with normal weight in an uneventful pregnancy. The mother was on 200 mg thyroxine due to hypothyroidism. The patient had normal developmental milestones with no associated physical abnormalities. The parents are first degree relatives and healthy, without any congenital anomalies. No other congenital disorder was known in the family (Figure 4).
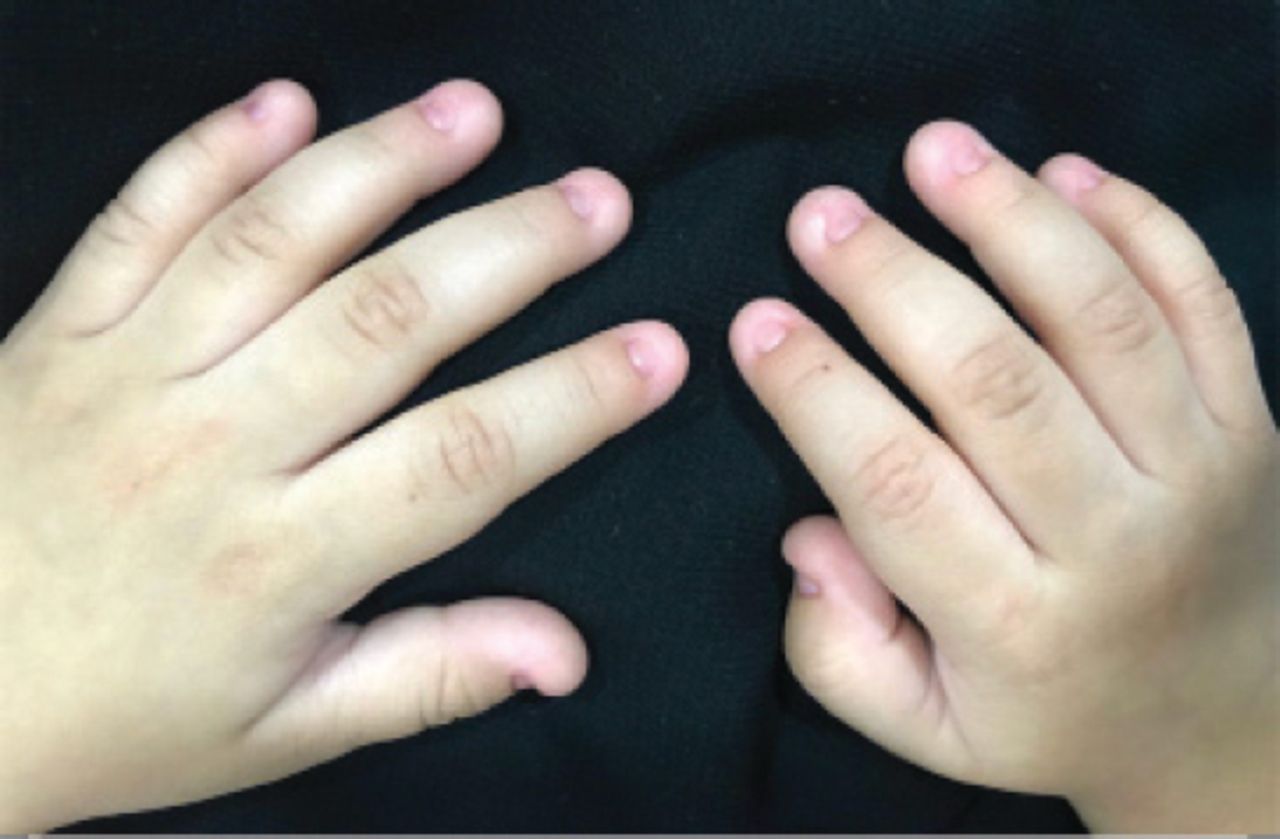
Total anonychia of all fingernails of patient 3.
Clinical findings
Upon examination, all the 3 patients exhibited absence of nail plates in hands and feet with normal nail beds, nail folds, and phalanges. The skin of the fingers and toes was normal. Systemic examination included teeth, hair, and mucous membranes and was normal. Sensory and motor functions of hands and feet were intact.
Diagnostic assessment
Plain x-ray examination of the hands and feet of all the patients revealed normal appearance of the bones (Figure 1 & 2). Rest of the skeletal survey was also normal. Visual acuity and pure tone audiometry were also normal. Based on all the observations, the 3 patients were diagnosed as anonychia congenita patients. As shown in the timeline table (Table 1).
Timeline of 3 causes with anonychia congenita.
Discussion
Anonychia congenita, an inherited non-syndromic nail disorder which is characterized by isolated abnormalities of the nails. There are 10 different types of “Nail disorder, Nonsyndromic Congenital”.4 Anonychia congenita is characterized by complete absence or severe hypoplasia of all nails of the fingers and toes with no associated physical or mental abnormality. It is of an autosomal recessive mode of inheritance with RSPO4 gene mutation mapped to chromosome 20p13 with frameshift splice site and missense mutations in exon 2.3,5,6 Congenital anonychia may be associated with hypoplastic or missing underlying phalanges and patella, teeth abnormalities, microcephaly, curved digits, single transverse palmar crease, bizarre flexural pigmentation, hair abnormalities, and mental retardation with sensorineural deafness.7 However, a rare form exists which is without any associated bony defect as in our patients. Besides, congenital anonychia may be associated with other ectodermal or mesodermal deformities including deafness, onychodystrophy, osteodystrophy, and mental retardation syndrome, epidermolysis bullosa, and Iso-Kikuchi syndrome. Drugs such as carbamazepine, phenytoin, warfarin, morphine, and trimethadione taken by the mother during the first and second trimesters of pregnancy can lead to nail deformities.8
The literature search has identified only 74 reports of anonychia congenita until 2017, most of which have Pakistani roots.9 Nevertheless, there are few other reported cases in Turkey, Russia, Great Britain, United States of America, Holland, Brazil, Iran, India, Kazakhstan, Lebanon, Kuwait, Finland, and in Germany.3,6,10-13 In KSA, it has been reported in a single patient and in 2 members of the same generation in another family.10,14 The 3 patients in this study had complete anonychia as an isolated and asymptomatic condition which marks them as one of the rare reported cases in Saudi Arabia, as its running in different generations of a single family.
Therefore, it is essential to screen those cases for visual, auditory, skeletal, dermatological and neurological disorders. Unfortunately, there is no medical or surgical treatment available, though artificial nails can be considered.
In conclusion, this is a case report of anonychia congenita affecting 3 members of one generation in an extended family with multiple affected generations of Arabic origin in KSA. Genetic analysis will be employed to confirm the diagnosis.
Acknowledgment
The authors gratefully acknowledge Editage (www.editage.com) for English language editing.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received October 25, 2019.
- Accepted December 26, 2019.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.