


Abstract
Proteus syndrome (PS) is a rare overgrowth disorder that presents with asymmetrical growth of the bone and fat tissues following a mosaic pattern mutation. The estimated worldwide incidence is approximately one in one million live births. Proteus syndrome causes disfigurement and psychological impact through its effects on somatic tissue. Due to its rarity and diversity of tissues involved, it represents a significant challenge to caregivers and multidisciplinary medical teams. Here, we report a Saudi girl, with a large left cervical mass discovered antenatally. This mass was identified as a growing cystic hygroma, and she had features of overgrowth and hemangiomas. Whole exome sequencing was negative from the blood lymphocytes and affected tissue sample. However, deletion duplication analysis from tissue shows a novel mosaic somatic mutation of the AKT1 gene. Somatic mutation remains an obstacle, and the geneticist has an essential role in its management, providing an established genetic diagnosis, prognosis, and family counselling.
Proteus syndrome (PS) is a rare medical condition that was first described in 1979 by Cohen and Hyden, and it was named Proteus after a Greek god, who was able to transform his body into multiple shapes.1 Proteus syndrome is characterized by overgrowth of many tissues that can be localized in a particular body segment or involve many areas. The affected tissues are derived from all 3 germ layers. The onset of overgrowth typically occurs in the first year of life. The skin, bones, and adipose tissue are commonly affected.2 The syndrome is a mosaic genetic syndrome that occurs de novo, with an estimated prevalence of less than one per one million.3 Proteus syndrome can be associated with bony defects, epidermal nevi, vascular tissue malformations, adipose tissue dyregulation, and lungs abnormalities. The presence of cerebriform connective tissue nevi is almost pathognomonic sign for PS. Nevi are most frequently seen on foot plantar surfaces, but have also been observed on other body parts. Approximately 10% of patients with PS have a history of epilepsy.4 Individuals with somatic mosaicism have 2 or more genotypically distinct populations of tissues that could be contained in different parts of the body. A molecular test of mosaicism is challenging for many reasons. First, mosaicism levels can differ widely and can be difficult to detect. Second, mosaicism might be specific or limited to a tissue. Sampling from multiple tissues within an individual might be needed for revealing mosaicism, which can severely hinder analysis due to the necessary and appropriate limitations of samplings. In the case of PS, the AKT1 somatic mutation is rarely present in the lymphocytes DNA.5 In this paper, we present the first described somatic novel duplication of the AKT1 gene. All previously described individuals with clinically-confirmed PS were caused by somatic mosaicism for the specific de novo pathogenic variant c.49G>A (p.Glu17Lys). We also describe the outcome of surgical intervention in this condition.
Case Report
Patient information
Our proband is a baby girl born at 35 weeks of pregnancy by cesarian section. Antenatally, there was an abnormal mass on the left side of the neck occupying the entire neck area and extending to the ear and shoulder. The parents are first cousins with no similar history in the extended family.
Clinical findings
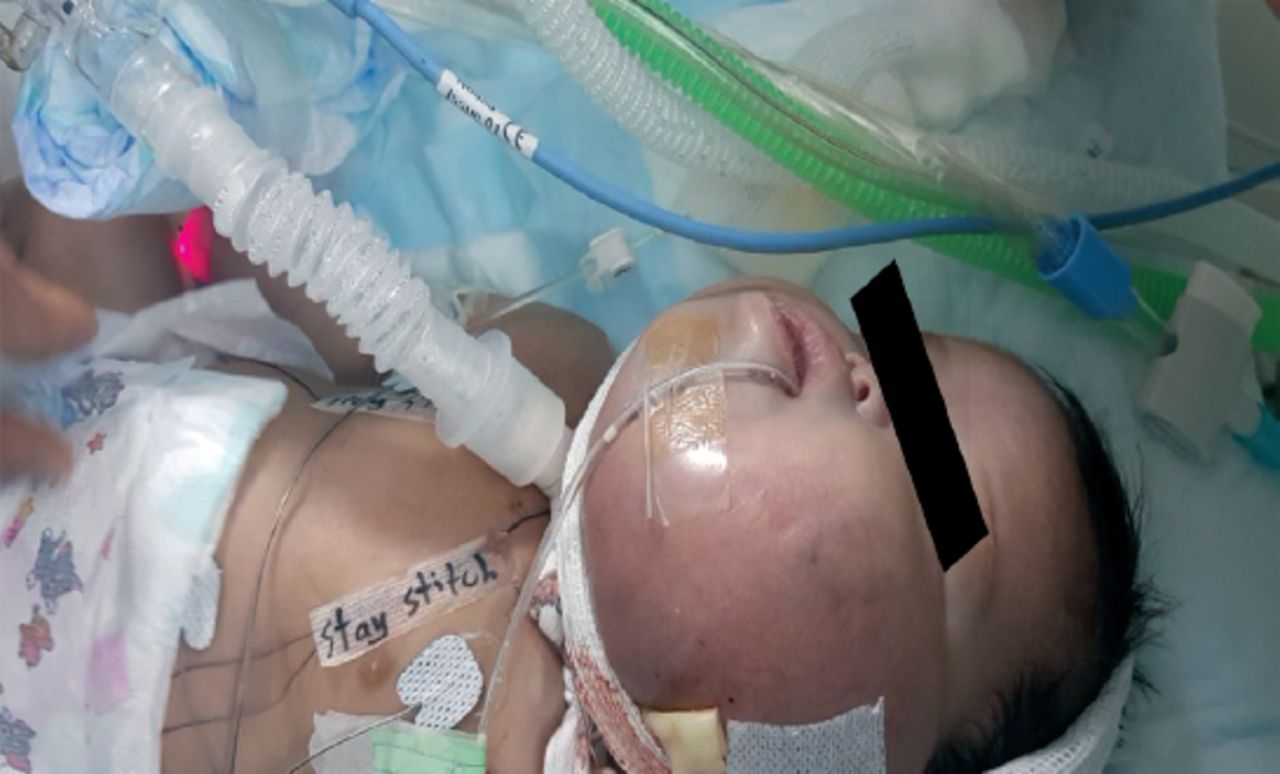
The planed EXIT procedure (ex utero intrapartum treatment) was conducted successfully. The baby was intubated with a size-3 tube, and a large neck mass was noted on the left side. There was an abnormal dark pigmentation over the mass and patches over the ipsilateral bodyside, with relatively larger digits of the same side.
Upon examination, the growth parameters were within normal limits: weight, 3 kg (25-50th percentile); head circumference, 34 cm (25-50th percentile); and length, 50 cm (50th percentile); vitally stable; head of normal size, with anterior fontanel and palpable suture lines; normal eye exam; no nystagmus; no cleft lip or palate. There was a large, cystic, multilocular mass in the neck (non-pulsating, compressible, trans-illuminated, 10×10 cm size, dark pigmented overlying skin, no bruit on auscultation) (Figure 1).

Left cheek showing large cystic hygroma.
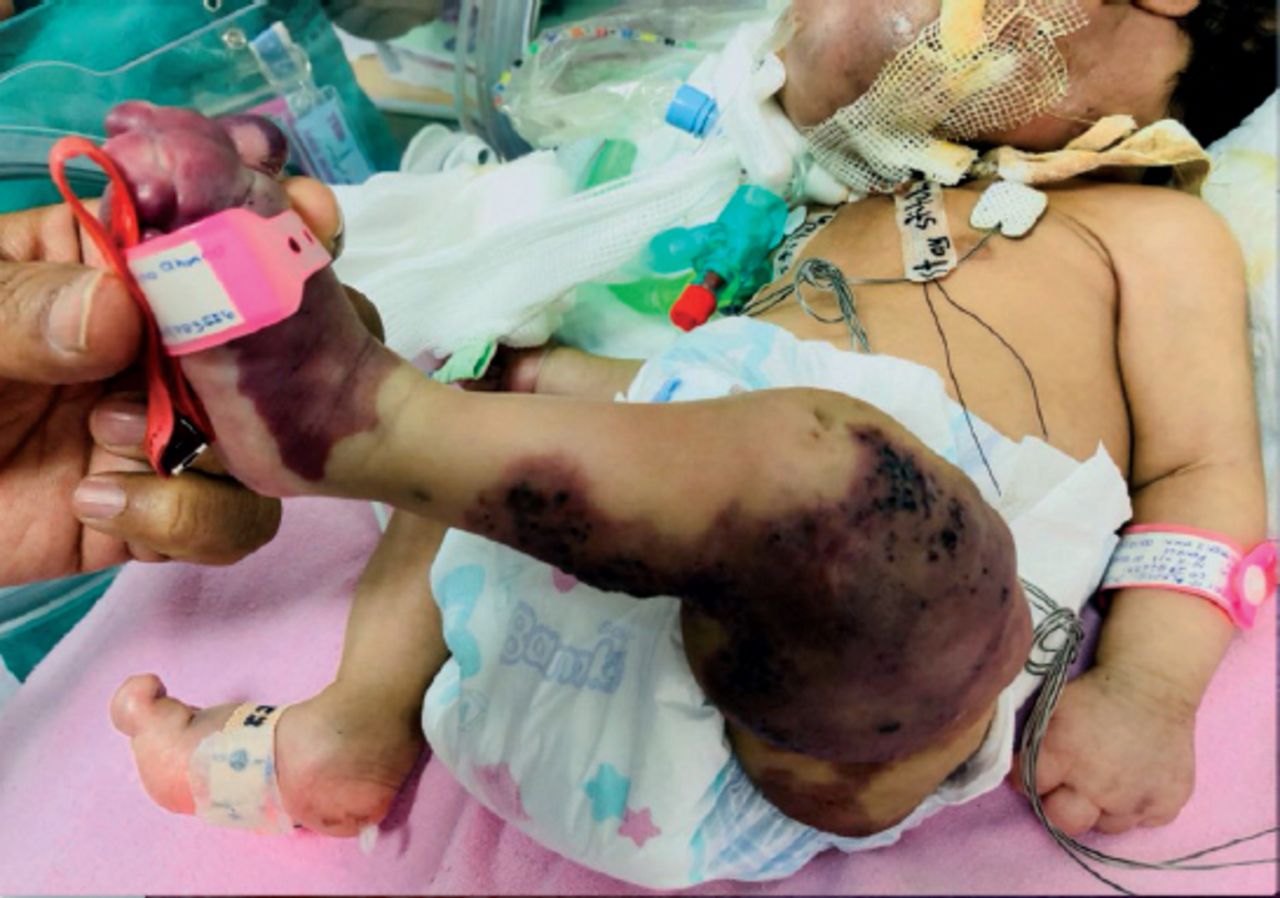
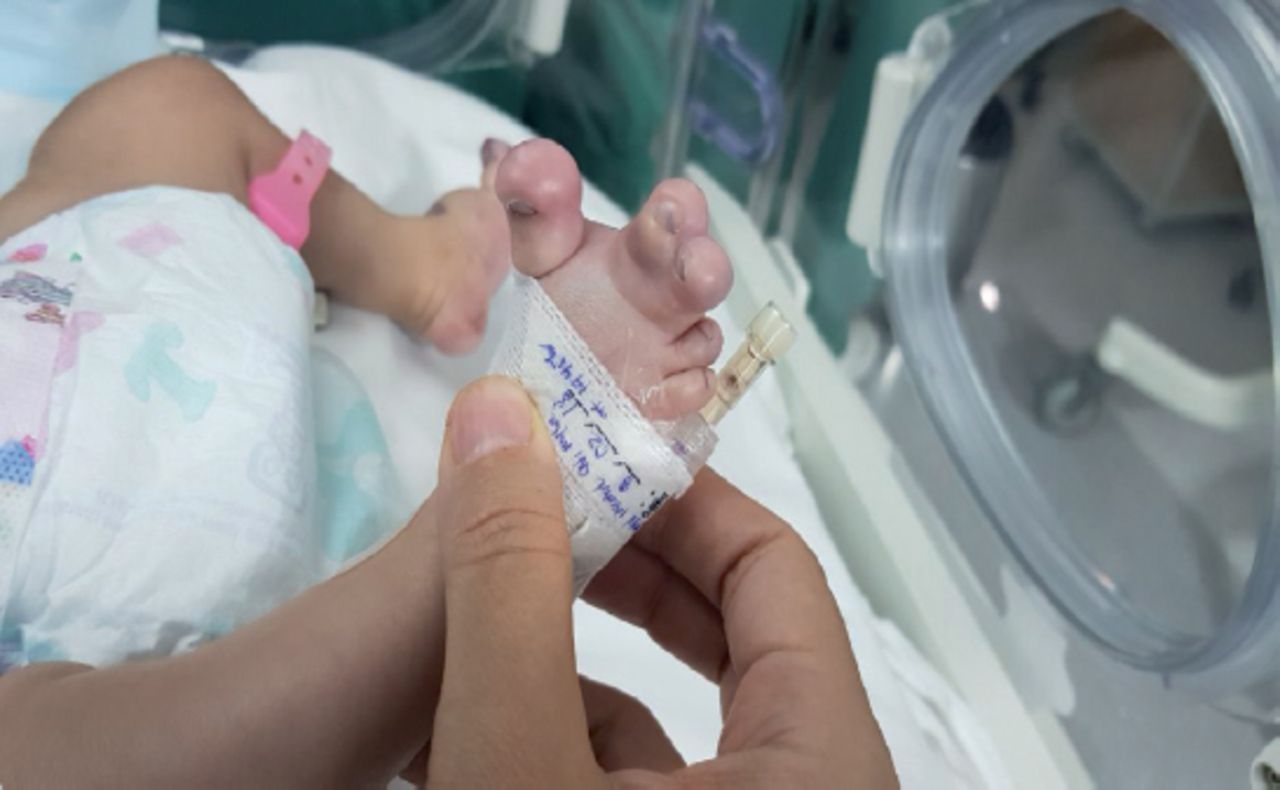
In the legs, there were deeply dark pigmented patches with verrucous texture-verrucous epidermal nevi, large size, and extending from the left mid-thigh to the foot with macrodactyly and syndactyly of the 2nd and third toes (Figures 2 & 3).

Left leg showing large epidermal nevi.

Left leg showing macro-syndactyly.
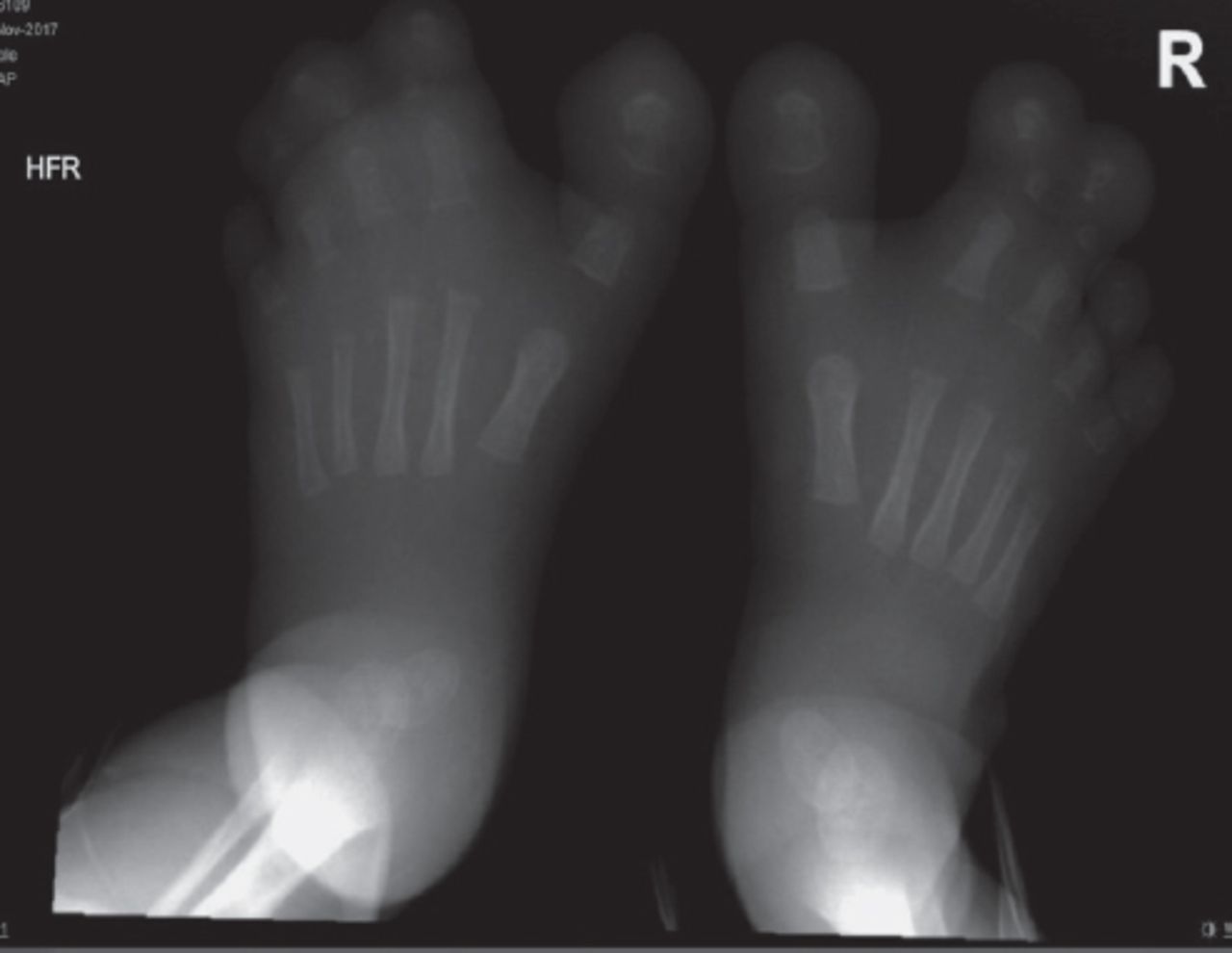
The skeletal survey revealed macrodactyly of the 2nd and 3rd toes, bilaterally. There was soft tissue syndactyly of the 2nd, 3rd, and 4th left toes and of the 2nd and 3rd right toes, and excessive soft tissue of the forefeet, bilaterally. We detected hypoplasia of the terminal phalanx of the left little toe (Figure 4). An abdominal ultrasound examination showed portal vein thrombosis.

X ray of the feet showing the macrodactyly.
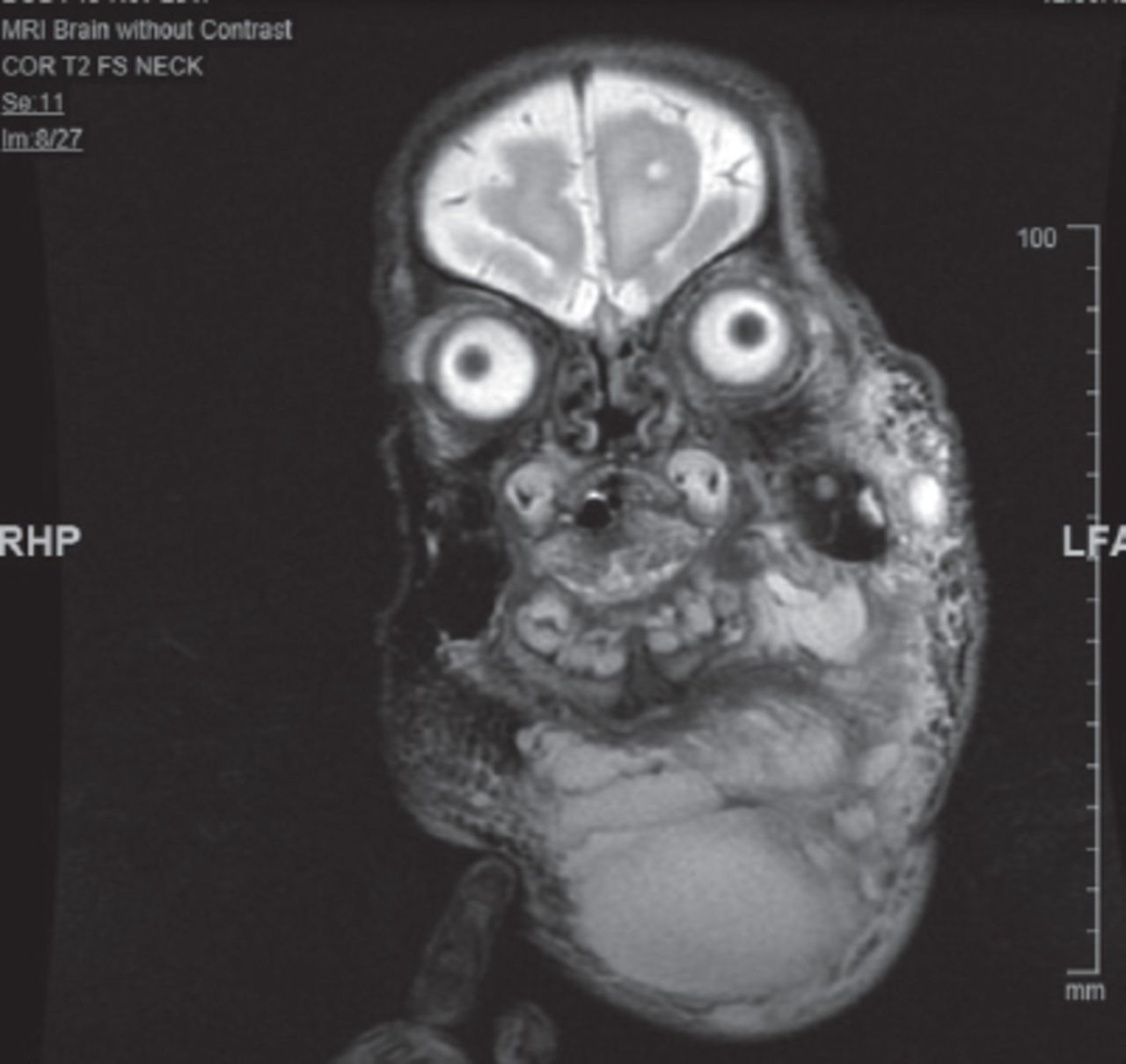
Brain and cervical magnetic resonance imaging (MRI) showed large mixed micro and macrocystic lymphatic malformation (hygroma) with suspicion of a small venous component (Figure 5). The pediatric otorhinolaryngology team, along with the interventional radiologist, were involved in the management of the lymphatic malformation.
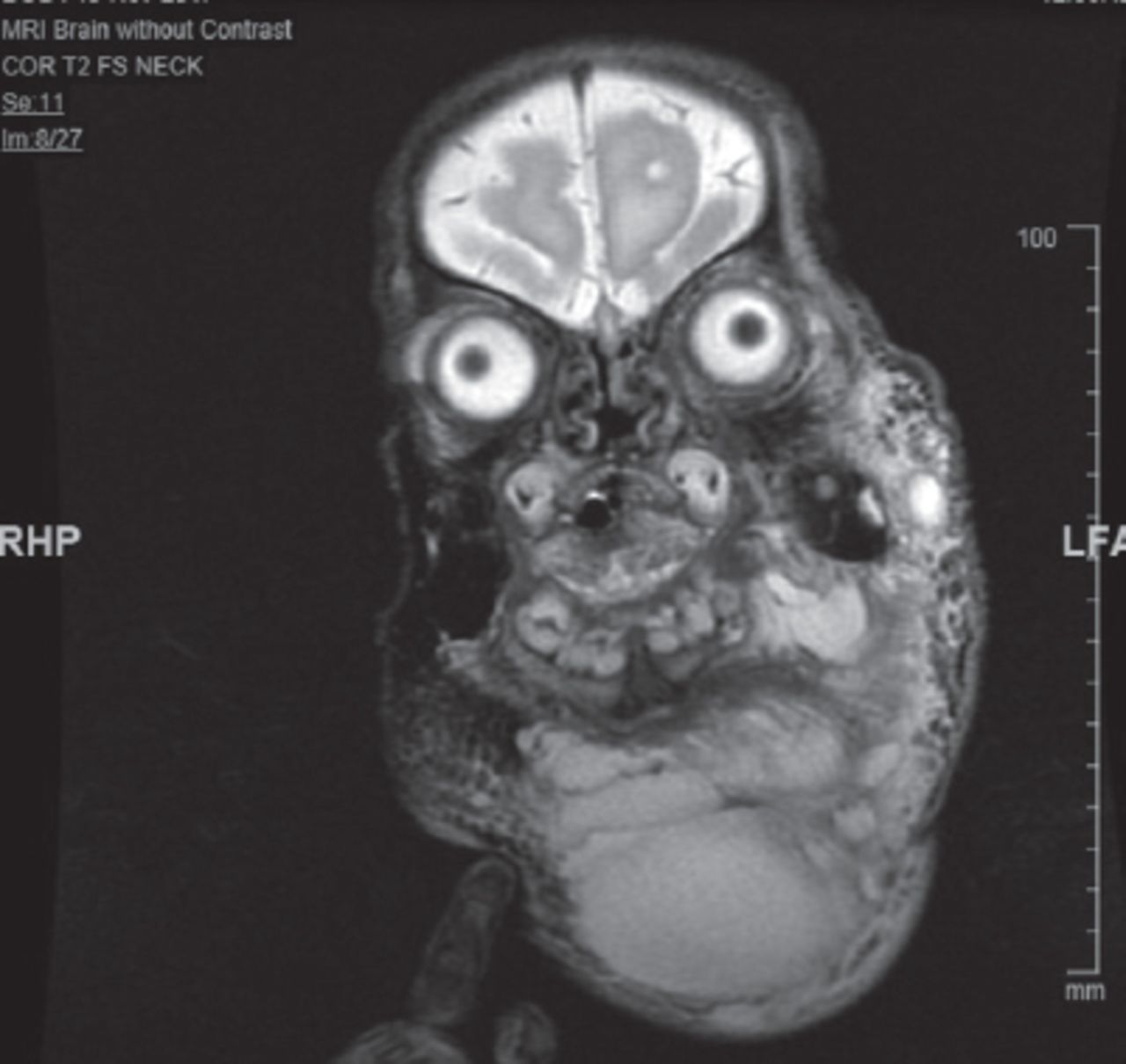
Magnetic resonance imaging of the head and neck showing the large multilocular cystic hygroma.
Therapeutic intervention
The pediatric otorhinolaryngology team decided to proceed with surgical excision and tracheostomy. The procedure was successful; however, there was a remnant of the disease mainly at the left parotid and the floor of the mouth. Furthermore, the patient underwent two sessions of sclerotherapy (using Belomycine) to control the remainder of the disease. The sclerotherapy was unsuccessful due to the nature of mixed micro-cystic diseases. The tracheostomy tube was accidentally obstructed, which caused the patient to develop severe anoxic encephalopathy and sadly pass away at 8 months old.
Diagnostic assessment
Clinically, there was a suspicion of PS because of the constellation of the phenotypic features. Whole-exome sequencing from the blood lymphocytes was negative. AKT1 gene sequencing was carried out using a skin biopsy of the affected tissue, but the result was negative. Next, an AKT gene duplication/deletion panel was performed, and the result was heterozygous duplication encompassing exon 3 to 15, which is novel and not previously detected in children with PS. The lab classified this variant as likely pathogenic class 2, according to the ACMG guidelines.
Follow-up and outcomes
The patient remained in the NICU for cystic hygroma management, unfortunately, she passed away due to uncontrolled epileptic encephalopathy and recurrent airway blockage by the large cystic hygroma despite two times of sclerotherapy. Do not resuscitate DNR status was discussed with the family and they agreed upon it.
Discussion
Proteus syndrome is an extremely rare disorder characterized by multiple tissues overgrowth, particularly bone and fat, vascular malformations, cerebriform lesions, or epidermal nevi. Due to its rarity and high variable phenotype, misdiagnosis of PS has been common before Biesecker first published the diagnostic criteria in 1999, which were updated by Turner in 2004.6 Patients must meet both the general criteria and the specific categorical criteria in order to be labelled with the diagnosis of PS. Our patient fulfilled these criteria (Table 2).
Patient clinical history.
Lindhurst et al5 have developed new criteria for PS diagnosis, which has more sensitivity than Turner criteria6 (Table 2).
General and specific categorical criteria in order to be labelled with the diagnosis of Proteus syndrome.
Guidelines for the evaluation and management of the patients with PS have been elaborated, including clinical photos, skeletal X-rays of the affected body areas, computerized tomography scans, MRI, and other analyses, such as dermatology, ENT, neurology, ophthalmology, and hematology. About 20% of PS patients die prematurely from a pulmonary embolism, postoperative complications, and pneumonia. The risk of a deep venous thrombosis has to be considered when managing PS patients. The advantages and risks of a surgical procedure must be thoroughly evaluated, and all precautions must be taken if an intervention is necessary.6 Lastly, this disorder is not inherited, and it is due to a de novo mutation in the AKT1 gene, which is acquired and shows a somatic pattern.
In conclusion, PS is a rare overgrowth syndrome characterized by somatic mutation of the AKT1 gene; in our case, it is a novel variant duplication of exon 3 to 15. Proteus syndrome can manifest as a medical emergency as it can compromise the airway. Its recurrence rate is near zero, as all cases show de novo mutations while the parents do not carry the mutation.
Acknowledgment
We would like to acknowledge the MRI Department of Pediatrics, Division of Medical Genetic, Prince Sultan Military Medical City, Riyadh, Kingdom of Saudi Arabia for the images and for their valuable contribution. All authors have given written permission to be acknowledged. The first and second author review the figures for clarity. Lastly, we would like to thank American Manuscript Editors (www.americanmanuscripteditors.com) for English language editing.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received October 13, 2020.
- Accepted December 6, 2020.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.