


Abstract
Objectives: To study childhood nephrolithiasis and nephrocalcinosis caused by metabolic disorders, distal renal tubular acidosis (dRTA), and familial hypomagnesemia, hypercalciuria, and nephrocalcinosis (FHHNC).
Methods: We retrospectively evaluated 86 children presented over 10 years (2011-2021), with nephrolithiasis (89%) and nephrocalcinosis (11%) caused by metabolic disorders (62%), FHHNC (21%), and dRTA (17%).
Results: The mean age at discovery was 72.7 months. The underlying metabolic etiologies included hyperoxaluria (38%), cystinuria (32%), hypercalciuria (24%), and hyperuricosuria (6%). Genetic testing was carried out for 23 patients. Hyperoxaluria was typically treated medically (75%). However, the majority progressed to end-stage kidney disease (ESKD). Most children with cystinuria, hypercalciuria, and hyperuricosuria required medical and surgical intervention. Patients with FHHNC typically presented with nephrocalcinosis. Genetic testing revealed Claudin-16 mutations in 7 children. Patients often progressed to stage II-IV chronic kidney disease (61%) and ESKD (6%). Patients with dRTA typically presented with nephrocalcinosis (80%), as well as poor weight gain and failure to thrive (86%), and medical treatment included sodium bicarbonate and potassium replacement. Despite nephrocalcinosis progression, most patients had normal renal function (53%), although the remaining 47% progressed to chronic kidney disease (none reached ESKD).
Conclusion: Childhood nephrolithiasis is mainly related to metabolic disorders and is associated with poor renal outcomes. Nephrocalcinosis and nephrolithiasis have poor outcomes when associated with FHHNC, while nephrocalcinosis associated with dRTA has relatively good renal outcomes.
Metabolic disorders are increasingly recognized as a cause of nephrolithiasis (renal stones) in children.1 Distal renal tubular acidosis (dRTA) is an important cause of nephrocalcinosis and to a lesser extent, nephrolithiasis.2 Nephrolithiasis is also commonly related to inherited defects in the thick ascending limb of the loop of Henle and connecting tubules.3 Altered claudin expression in the thick ascending loop tight junctions influences calcium homeostasis, with mutations in the genes encoding Claudin-16 (CLDN16) or Claudin-19 potentially causing familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC), while gain-of-function mutations affecting Claudin-14 can cause kidney nephrolithiasis.4 Environmental factors are the main cause of adult nephrolithiasis, although genetic or metabolic disorders are the main causes of childhood nephrocalcinosis and urolithiasis.5,6
Early diagnosis may prevent or minimize serious long-term complications of these diseases. The diagnosis is based on imaging (ultrasonography, plain radiography, or non-enhanced computed tomography [CT]) used to locate the calculi and identify urinary tract anomalies or acute obstruction caused by the nephrolithiasis. Diagnostic evaluation of the underlying causes is also mandatory, which should include urine testing, genetic testing, and stone analysis.6 The management of pediatric nephrolithiasis includes medical and surgical treatments that aim to eliminate the burden of kidney stones and prevent recurrence.7 However, there is a paucity of studies regarding childhood nephrolithiasis, especially patients from Saudi Arabia. The present study evaluated the clinical features and outcomes of nephrolithiasis and nephrocalcinosis caused by metabolic disorders (hypercalciuria, hyperoxaluria, cystinuria, and hyperuricosuria), dRTA, and FHHNC at 2 tertiary centers in Saudi Arabia.
Methods
The retrospective study included 86 children and adolescents with nephrolithiasis and nephrocalcinosis who were treated at King Abdulaziz University (KAU), Jeddah, and King Saud University Medical City Hospital, Riyadh, Saudi Arabia, between January 2010 and December 2018.
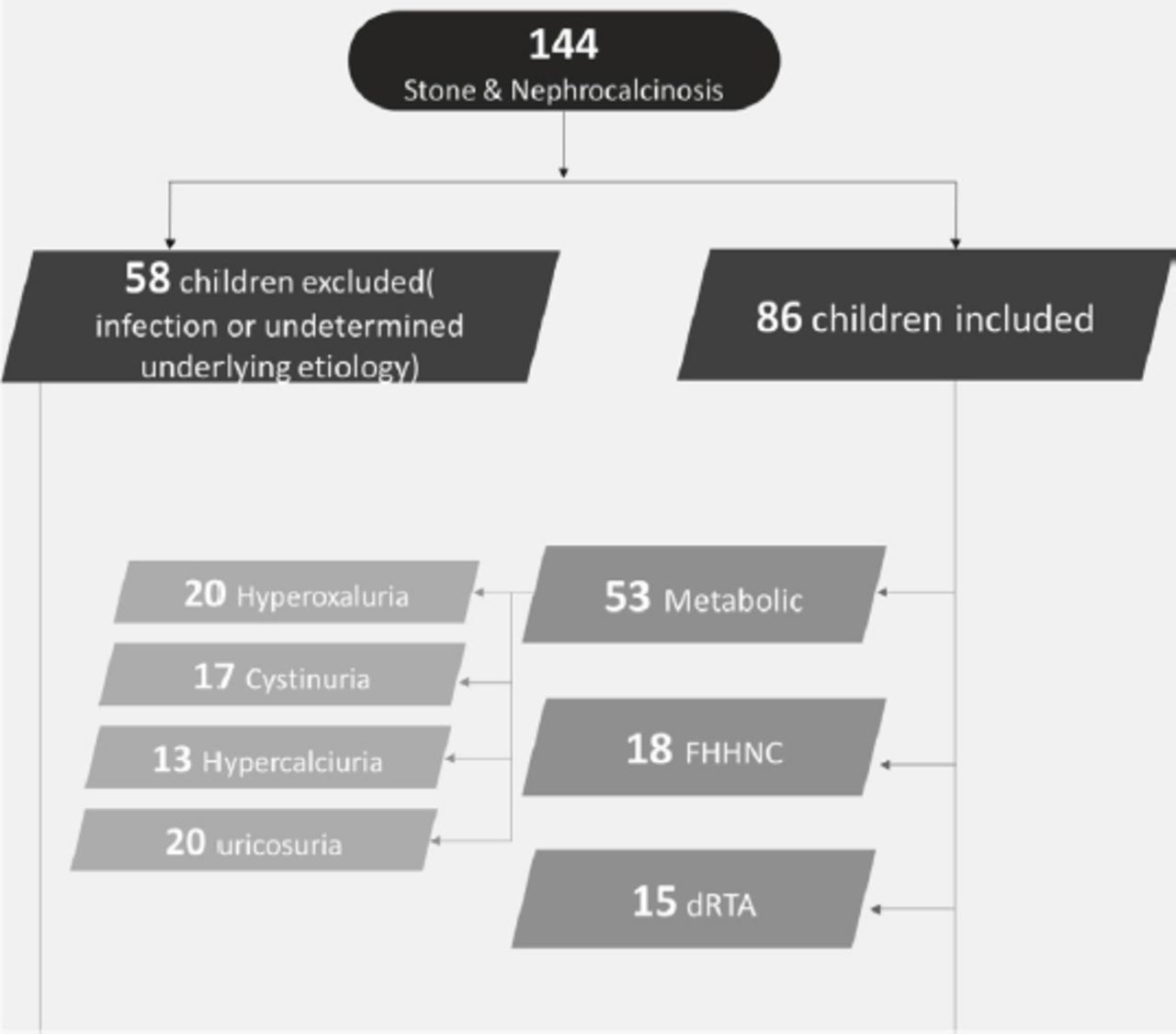
Electronic medical records were reviewed. Patients were considered eligible (inclusion criteria) if they were <18 years old and had a radiological diagnosis of renal stones or nephrocalcinosis due to metabolic renal diseases and renal tubulopathy. Patients were excluded if they had nephrolithiasis or nephrocalcinosis that was related to infection and congenital renal anomalies (21 patients) or undetermined causes (37 patients).
The patients’ clinical and metabolic features were reviewed based on clinical findings, urine testing, genetic testing, and stone analysis. The treatment involved medical therapy and lithotripsy or surgical removal depending on the patient’s characteristics. Thus, patients were grouped according to whether they required medical treatment alone, medical treatment and lithotripsy or medical and surgical treatment. Figure 1 shows how patients were assigned to the different groups.

- A flow diagram showing how patients were assigned to the different groups. FHHNC: familial hypomagnesemia with hypercalciuria and nephrocalcinosis, dRTA: distal renal tubular acidosis
Nephrolithiasis was identified based on the confirmed presence of a stone in 2 images (excluding artifacts) or if stone analysis results were available after surgical removal or spontaneous passage of a stone. Renal ultrasound, x-ray or CT scans were used for diagnostic imaging. Nephrocalcinosis was identified based on medullary calcification without shadowing in a part of the renal medulla. Ultrasonography or CT was used to evaluate faint calcifications. Nephrocalcinosis was classified based on the imaging findings (ultrasonography, radiography, and CT) as mild (early hyperechogenicity at the periphery of the pyramids), moderate (diffuse hyperechoic pyramids), and severe (clumps of renal pyramids).8
The study was approved by the Ethical Committees of KAU, Faculty of Medicine, Jeddah, and King Saud University Medical City Hospital, Riyadh, Saudi Arabia, and was carried out in accordance with the Declaration of Helsinki.
The patients’ records were searched to collect data regarding age at presentation, gender, presenting symptoms, creatinine concentration, estimated glomerular filtration rate (eGFR) at presentation, and history of consanguinity or family history of renal disease. The different groups were compared in terms of the dominant findings (nephrolithiasis versus nephrocalcinosis) and stone characteristics (number, location, and composition). The metabolic workup for nephrolithiasis was carried out using a spot urine sample and interpreted as the solute/creatinine ratio 9, with duplicate testing to confirm the presence of hypercalciuria, hyperoxaluria, cystinuria, and hypocitraturia or hyperuricosuria (Appendix 1). In addition, hypermagnesiuria was identified based on the mean fractional excretion of magnesium (FEMg%) and hyperphosphaturia was identified based on the mean transtubular reabsorption of phosphate. Urine test results were reviewed to identify evidence of a urinary tract infection (UTI) based on the presence of red blood cells, white blood cells, and bacteria.
Blood test results were reviewed to evaluate kidney function, potential of hydrogen (pH), and serum concentrations of sodium, potassium, chloride, bicarbonate, calcium, phosphate, magnesium, and uric acid. The results were used to identify metabolic acidosis/alkalosis, hypo/hypernatremia, hypo/hyperkalemia, hypo/hyperchloremia, hypo/hypercalcemia, and hypo/hyperphosphatemia. Hypernatremia was defined as a serum sodium concentration of >145mmol/L and hyponatremia was defined as a serum sodium concentration of <135mmol/L. Hyperkalemia was defined as a serum potassium concentration of >5.5mEq/L in children or >6mEq/L in neonates, and hypokalemia was defined as a potassium concentration of <3.5mEq/L. Hypercalcemia was defined as a serum calcium concentration of >2.6mmol/L and hyperphosphatemia was defined as a serum phosphate concentration of >1.58mmol/L. Metabolic acidosis was defined as a pH value of <7.35, plus a serum bicarbonate concentration of <18mEq/L, while metabolic alkalosis was defined as a pH value of >7.45, plus a serum bicarbonate concentration of >25mEq/L. The eGFR was calculated using the Schwartz formula to evaluate chronic kidney disease (CKD) progression.10
Genetic tests were carried out for 23 patients using Whole Exome Sequencing through CentoXome® Solo (including next generation sequencing [NGS]-based copy number variation analysis), which could be requested through private providers (Centogene or Perkins) or through collaboration with genetic centers, such as Harvard Medical School, Boston, United States. The copy number variation detection software used in this study has a sensitivity of more than 95% for all homozygous/hemizygous and mitochondrial deletions, as well as heterozygous deletions/duplications and homozygous/hemizygous duplications spanning at least 3 consecutive exons. These laboratories have established stringent quality criteria and validation processes for variants detected by NGS. Variants with low quality or unclear zygosity were confirmed by orthogonal methods. Consequently, a specificity of >99.9% for all reported variants is warranted. Familial carrier testing for known mutations was always available upon request of the treating physician.
Statistical analysis
Outcomes for the various groups were evaluated according to calcification and renal function. All analyses were performed using STATA software (release 12; StataCorp LP, College Station, TX). Categorical variables were presented as numbers (percentages) and analyzed using the Chi-squared or Fisher’s exact test. Continuous variables were reported as means (95% confidence interval [CI] of the mean) and analyzed using Student’s t-test. A p-value of <0.05 was considered significant.
Results
We identified 86 eligible patients with nephrolithiasis or nephrocalcinosis, which were related to metabolic disorders (61.6%), FHHNC (20.9%), or dRTA (17.5%). All 3 groups had a similar mean age (70-73 months) at presentation (Table 1). The majority of patients were male in the metabolic and dRTA groups (60%) and female in the FHHNC group (67%). The metabolic group had higher mean creatinine concentration at presentation compared to the dRTA and FHHNC groups (103.7 µmol/L vs. 72.7 µmol/L vs. 71.8 µmol/L). A family history of renal disease was more common among patients with FHHNC (Table 1).
- Patients’ characteristics according to disease group.
The metabolic disorders included hyperoxaluria (38%), cystinuria (32%), hypercalciuria (24%), and hyperuricosuria (6%). The average age at presentation was older for hyperoxaluria and cystinuria (70-91 months) than for hypercalciuria and hyperuricosuria (59-56 months). Furthermore, the creatinine concentration at presentation was higher for patients with hyperoxaluria and cystinuria than for patients with hypercalciuria and hyperuricosuria (178.3 µmol/L vs. 77 µmol/L vs. 37.2 µmol/L vs. 44 µmol/L). A history of consanguinity was common, especially for patients with hyperoxaluria and cystinuria (Tables 2&3). Loin pain, UTI, failure to thrive, and hematuria were the most common presentations. Most patients had multiple renal stones (88.7%) and the rest had nephrocalcinosis (11.3%).
- Patients’ characteristics according to metabolic group.
- Laboratory and radiological findings according to metabolic group.
Renal stones were observed in all patients with hyperuricosuria and cystinuria, as well as in most patients with hypercalciuria and hyperoxaluria (85-80%), while bladder stones (urolithiasis) were common in patients with cystinuria (94.1%). Nephrocalcinosis was observed in patients with hyperoxaluria (20%) and hypercalciuria (15.4%), which was graded as severe (grade III, 50% of cases), moderate (grade II, 33.3% of cases), and mild (grade I, 16.7% of cases). Stone analysis results were available for 4 of 13 patients (30.7%) with hypercalciuria (3 patients had calcium carbonate and one patient had calcium oxalate), 33% had hyperuricosuria, 62.5% had hyperoxaluria (calcium oxalate), and 94% had cystinuria. Genetic testing revealed mutations in alanine-glyoxylate and serine-pyruvate aminotransferase (AGXT) in 11 of 20 patients with hyperoxaluria and in solute carrier family 3 member 1 (SLC3A1) in 3 of 17 patients with cystinuria (Table 4).
- Details of genetic analysis.
Most patients received medical therapy, especially in cases of hyperoxaluria (47%). Medical and surgical intervention was required for most patients with hyperuricosuria, hypercalciuria, and cystinuria. However, radiological follow-up revealed nephrolithiasis and nephrocalcinosis worsening or progression in 68% of children with hyperoxaluria and 67% of children with hyperuricosuria. Some patients (30%) with hyperoxaluria progressed to end-stage kidney disease (ESKD), while almost one third of patients with hypercalciuria and cystinuria progressed to stage II-IV CKD.
A diagnosis of FHHNC was made for 18 (20.9%) patients, who had variable presentations that involved recurrent UTI, failure to thrive, hematuria, and accidental discovery during routine investigations (Table 5). These patients had elevated urine calcium concentrations (mean urine calcium/creatinine ratio: 2.43 mmol/mmol, 95% CI: [1.62-3.24]), hypermagnesuria (mean FEMg%: 15.46%, 95% CI: [11.09-19.82]), significant alkalosis (mean pH: 7.48, 95% CI: [7.46-7.50], mean serum bicarbonate: 29.83 mEq/L, 95% CI: [29.31-30.35] mEq/L), and hypomagnesemia (mean serum magnesium: 0.54 mEq/L, 95% CI: [0.50-0.59 mEq/L]).
- Clinical presentation and chemistry results among patients with FHHNC and dRTA.
Genetic testing had been performed on 7 patients, which identified pathogenic variant mutations in CLDN16.
Consanguinity was noted for all of these cases. These patients presented with radiological evidence of nephrocalcinosis, which was considered either moderate (grade II, 55.6%), severe (grade III, 33.3%), or mild (grade I, 11.1%). Most patients with FHHNC received medical supportive therapy for CKD. Radiological progression of nephrocalcinosis was observed in 94.4% of cases (no change for 5.6%), which resulted in stage II-IV CKD (61.1%), ESKD (5.6%), or normal renal function throughout follow-up (33.3%)
A diagnosis of dRTA was carried out for 15 (10%) patients, who generally presented with poor weight gain and failure to thrive (86%), although accidental discovery was noted for 14% of these patients (Table 4). These patients typically had hypokalemia (mean potassium: 3.14 mEq/L, 95% CI: [2.9-3.3 mEq/L]), hyperchloremia (mean chloride: 109.5 mEq/L, 95% CI: [105.5-113.54 mEq/L]), metabolic acidosis (mean pH: 7.25, 95% CI: [7.21-7.30], mean serum bicarbonate: 17.8 mEq/L, 95% CI: [16.2-19.38 mEq/L]), and borderline low trans-tubular phosphate reabsorption (mean: 83.6%, 95% CI: [76.9-90.4]; Table 5). Nephrocalcinosis was present in 80% of these patients, which was considered either moderate (grade II, 66.7%) or mild (grade I, 33.3%). Nephrolithiasis was observed in 20% of these patients, which involved a single stone (66.7%) or multiple stones (33.3%) located in the renal pelvis or ureters, but not in the bladder.
The patients received sodium bicarbonate and potassium replacement in addition to supportive treatment for CKD. Radiological evidence of nephrocalcinosis worsening was observed in 80% of these patients (no change in 11.3%). Interventional or surgical treatment was required for 6.7% of the patients with dRTA. Most patients maintained normal kidney function (53.3%), although progression to stage II-IV CKD was observed in 46.7% of the patients. No patients with dRTA progressed to ESKD during follow-up.
Discussion
This study revealed that metabolic disorders were the main causes of childhood nephrolithiasis, while FHHNC and dRTA were the main causes of nephrocalcinosis. These findings are similar to those from previous reports and support previous recommendations that evaluated children with nephrolithiasis and nephrocalcinosis for underlying hereditary diseases.1,6,11 Similar to previous reports, we were able to identify the underlying etiology in most cases (75%).6,12 Furthermore, patients in all 3 groups commonly presented with loin pain, hematuria, or UTI, which suggested that a suspicion of nephrolithiasis and nephrocalcinosis was warranted for children with these symptoms.13 During the last 3 decades, the incidence of nephrolithiasis has increased to approximately 50 cases per 100,000 adolescents, and many reports have indicated that the underlying causes have shifted from predominantly infectious diseases to metabolic disorders.14,15 However, we observed that the children had variable symptoms at presentation, and some children even had no symptoms, which supports the generally non-specific presentation of childhood nephrolithiasis and the relatively small proportion of patients who present with classic unilateral colicky flank pain.16 Nevertheless, a history of consanguinity was common in all 3 of our groups, which highlights the contribution of genetic factors. Genetic test results were carried out for 23 (27%) children and there are many recent reports regarding the importance of genetic testing when diagnosing nephrolithiasis.12,17,18
Metabolic disorders were the main underlying etiology in this study, which agrees with findings from previous studies.19,20 We also observed that male patients were more common, which agrees with some reports, although other studies have indicated a relatively equal distribution of male and female patients.15 More than one-third of the metabolic group had hyperoxaluria, which was confirmed by genetic testing in >50% of those cases. In contrast, hypercalciuria was reported as the most common underlying cause in a Turkish study and other older studies.1,15,19 Increased urine oxalate excretion may be caused by increased oxalate absorption, excessive intake of oxalate precursors, or rarely by an inherited metabolic disorder (primary hyperoxaluria [PH]).19 Type I and type II PH are relatively rare autosomal recessive disorders that involve endogenous oxalate over-production by the liver and result in excessive urinary oxalate excretion with resultant nephrocalcinosis and nephrolithiasis leading to CKD and ESKD, which we observed in this study. Furthermore, 55% of children with hyperoxaluria had mutations in the AGXT gene, which causes type I PH via a functional defect in alanine-glyoxylate aminotransferase (a hepatic peroxisomal enzyme). The high proportion of primary hyperoxaluria among our patients could be related to the commonness of consanguinity in Saudi Arabia. We also observed high creatinine concentrations at presentation, which could be related to the fact that most patients developed type I PH.
The second most common metabolic disorder was cystinuria (32%), which differs from previous reports, as it was not considered a common cause of childhood nephrolithiasis.16,19,21 This autosomal recessive disorder of renal tubular transport is associated with failure of the renal tubules to reabsorb 4 basic amino acids (cysteine, ornithine, lysine, and arginine) and causes lifelong recurrent nephrolithiasis.16,21 Most children with cystinuria had bladder stones, which has not been previously reported as a characteristic of cystinuria. Furthermore, most patients were diagnosed via stone analysis, and genetic analysis was only available for 3 (18%) children, which identified SLC3A1 mutations. We have also previously reported a broad spectrum of 12 SLC3A1 variants (including 5 novel variants) in children with cystinuria from Saudi Arabia.22 Most of these patients required a combination of medical and surgical therapy, which provided good kidney function outcomes.22
In this study, approximately 25% of the metabolic group had hypercalciuria and presented with nephrocalcinosis and nephrolithiasis. Hypercalciuria is considered a common metabolic cause of childhood nephrolithiasis and nephrocalcinosis, and is usually idiopathic with a good prognosis in terms of renal function.1,16,19 We included only idiopathic cases in this group and the lower proportion of these cases (vs. previous studies) was likely related to our classification of FHHNC as a separate etiological group. A total of 3 patients had uric acid nephrolithiasis and presented mainly with loin pain, although this was not associated with hyperuricemia. Uric acid stones are a rare case of childhood nephrolithiasis, which could be related to hyperuricosuria-associated significant hyperuricemia caused by disordered purine metabolism, lymphoproliferative disorders, and polycythemia.16 Hyperuricosuria can be caused by excessive purine intake (animal protein, anchovies, and mussels), hemolysis, uricosuric medications (probenecid, salicylates, and losartan), cyanotic congenital heart disease, melamine toxicity, and familial causes (typically idiopathic). Hereditary renal hypouricemia is characterized by low serum uric acid concentrations, hyperuricosuria, nephrolithiasis, and exercise-induced acute kidney injury, with rare cases involving mutations in the SLC22A12 or SLC2A9 genes.15 Unfortunately, we did not test for hypocitraturia, which could be idiopathic in most cases, but could also be related to a ketogenic diet, certain medications (topiramate, zonisamide, and acetazolamide), dRTA, and chronic diarrhea. An incomplete form of dRTA can occur in the absence of overt systemic acidosis or hypokalemia. A diet rich in animal protein, but low in vegetable fiber and potassium, seems to promote lower citrate excretion.15,23
Approximately 21% of our patients had FHHNC as the underlying etiology, which is considered an important cause of nephrocalcinosis, CKD, and ESKD.24 The diagnosis can be based on laboratory and radiological findings, as well as genetic testing, which confirmed the diagnosis in 39% of our cases. As an autosomal recessive tubular disorder, FHHNC is caused by CLDN16 mutations that lead to partial or complete function loss of claudin-16 (the renal tight junction protein), which determines the degree of CKD progression.25
Approximately 18% had dRTA as the underlying etiology, which can be diagnosed based on clinical, laboratory, and radiological findings. Most of these patients had nephrocalcinosis, although 20% had nephrolithiasis, which agrees with a previous report.2 The causes of dRTA include genetic factors, autoimmunity, or some medications. In children, it is mainly hereditary and caused by autosomal recessive mutations (ATP6V0A4, ATP6V1B1, FOXI1, and WDR72), as well as autosomal dominant or autosomal recessive SLC4A1 mutations.26 The majority of our patients in this group had preserved renal function, which was similar to previous reports.27
Study limitations
Its retrospective nature and the relatively small number of subjects. However, the findings provided information regarding a rare condition among children, with limited previously published data from Saudi Arabia. Because of the retrospective nature of this study, we could only refer to the progression of CKD as a quantitative analysis of glomerular filtration rate reduction from baseline at the time of data review. The frequency of follow-up visits, the adherence of familial visits, and the management provided were not assessed in all patients. Therefore, we were unable to get a more accurate description of CKD in relation to time.
In conclusion, we identified the etiology underlying nephrolithiasis or nephrocalcinosis for most of the children in this study. Metabolic disorders were the main underlying cause and were associated with relatively poor renal function outcomes. Furthermore, FHHNC was associated with nephrocalcinosis and nephrolithiasis, and these patients also had a poor prognosis. In contrast, dRTA was associated with nephrocalcinosis and good renal function outcomes. These findings may help guide the diagnosis in chilhood nephrolithiasis and nephrocalcinosis.
Acknowledgment
The authors gratefully acknowledge Editage (www.editage.com) for English language editing.
Appendix 1 - Cut-off level (95th centile) of solute: creatine ratio in relation to age group.
Footnotes
Disclosure. This study was funded by the Deanship of Scientific Research at the King Abdulaziz University, Jeddah, Saudi Arabia (G: 257-140-1439).
- Received September 11, 2021.
- Accepted December 7, 2021.
- Copyright: © Saudi Medical Journal
This is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
References
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.