


Abstract
Objectives: To develop a candidate vaccine aginst the Sphingobacterium spiritivorum.
Methods: Since there is currently no vaccine against this pathogen, we employed in-silico methods to extensively explore the outer membrane toxin-producing proteins found specifically in S. spiritivorum to forecast a multi-epitope chimeric vaccine design. This computational study was conducted in Saudi Arabia in 2022 (study design: computational; ethical approval not applicable).
Results: TThe vaccine peptide comprises multiple linear and conformational B-cell epitopes, which have the potential to elicit humoral immunity. Projected B-cell- derived T-cell epitopes for outer membrane proteins are present in the produced protein. The docking and molecular dynamic simulation results indicating that the chimeric vaccine had adequate binding stability with TLR-4. Following the immunological simulation, significant levels of immune cell expression were observed as immunoglobulin (Ig) M and IgG, IgM, IgM1, and IgM2, and independently IgG1 and IgG2.
Conclusion: The developed vaccine candidate is suitable for further testing and can assist experimental vaccinologists in developing an effective vaccine against S. spiritivorum.
Gram-negative environmental bacilli belonging to the genus Sphingobacterium are the only kind of Sphingobacterium bacteria that are ever associated with human infections. Two species of Sphingobacterium, Sphingobacterium multivorum and Sphingobacterium spiritivorum (both of which were earlier categorised as Flavobacterium spp.), have been sporadically related to bacteremia, peritonitis, and persistent respiratory infections.1,2 These links have been seen in some persons who have symptomatic underlying illnesses. Sphingobacterium are most frequently found in soil, vegetation, and water that has been standing for an extended period of time. Even though they are classified as opportunistic infections, they are capable of infecting hosts with a healthy immune system. There have already been reports of 4 individuals suffering from a sickness caused by S. spiritivorum. Cellulitis was implicated in 2 of these cases, extrinsic allergic alveolitis was involved in one case, and the most recent example featured septicemia infecting a patient with acute myeloid leukaemia.3,4 Patient with leukemia died; all other instances were treated with antibiotics and recovered. The source of infection in the case of the patient with extrinsic allergic alveolitis was identified as a bacterial strain that was isolated from the patient’s sputum and came from a water reservoir inside his steam iron.5
In the past, antibiotic therapy with cephalosporins or a combination of penicillin or ciprofloxacin was the primary treatment. The majority of Sphingobacterium are naturally resistant to polymyxin B and aminoglycosides.6 Several resource tests for antibiotic susceptibility show that Sphingobacterium is susceptible to quinolones and trimethoprim-sulfamethoxazole, with S. spiritivorum additionally being susceptible to carbapenems and certain cephalosporins.4,7 Furthermore, data indicate that implantable dialysis catheters always include inherent and unavoidable hazards; nevertheless, completely implanted devices have been proven to result in fewer infectious problems.8 Traditional vaccine development approaches are costly, time-consuming, and rarely produce successful outcomes.9,10 Recent breakthroughs in bioinformatics, immunoinformatics, and structural vaccinomics have transformed antigen screening. Reverse vaccinology, a novel method of vaccine development, was developed by combining these 2 approaches and is now used for vaccine development against numerous bacterial and viral diseases.11,12 A multi-epitope vaccine was recently proved to be more effective and protective against infectious pathogens than a single epitope-based vaccine.13
In this study, a reverse vaccinology strategy was taken in order to uncover prospective vaccine candidates by analyzing the whole proteome of Sphingobacterium spiritivorum. This methodology was chosen not only due to its many benefits, but also due to the fact that it chooses candidates from extracellular and outer membrane proteins based on whether or not they have shown encouraging outcomes in previous research utilizing animal models.14 In addition, T and B cell epitope mapping was performed on the filtered proteins. The top-ranked epitopes were subsequently used to design a multi-epitope vaccine, which was then examined for its structural and immunological characteristics in order to support its recognition as a promising vaccine candidate against S. spiritivorum. Several lines of the in-computer study point to the possibility that the anticipated vaccine candidate could be a viable treatment for S. spiritivorum.15,16
Methods
Proteome extraction and subcellular localization process
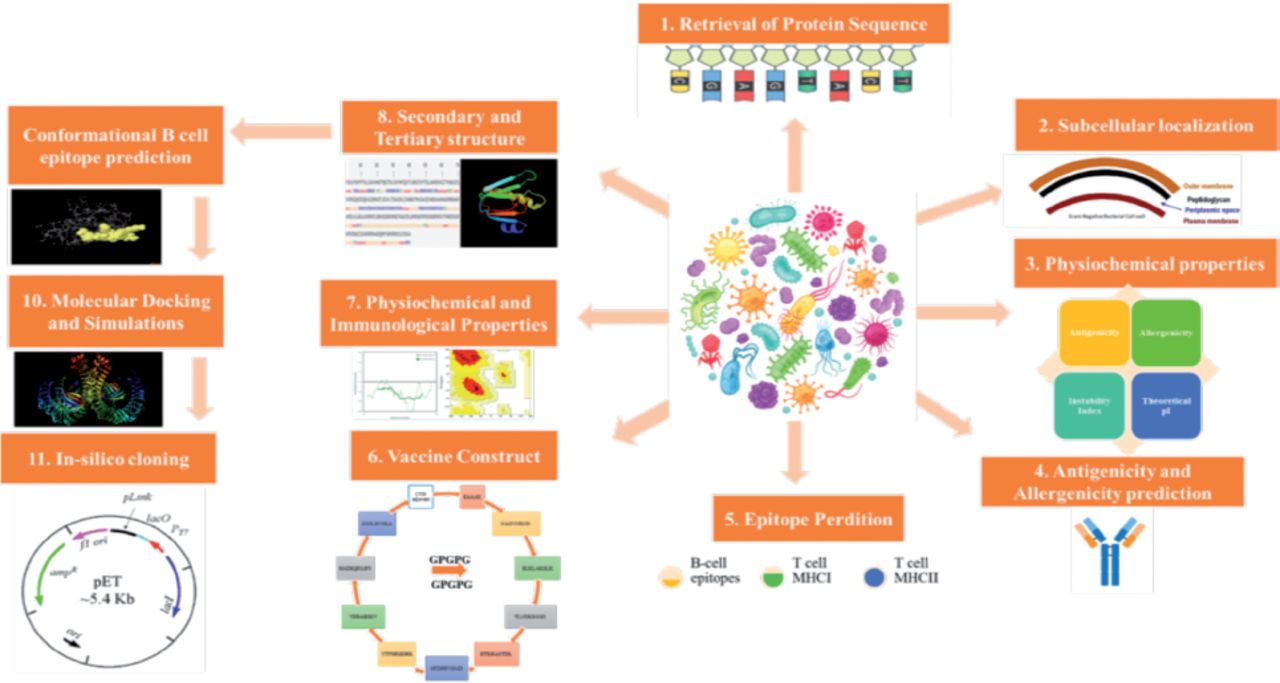
The National Center for Biotechnology Information (NCBI) provided proteome information for each strain of S. spiritivorum that had its whole genome sequenced.17 Core proteomes were assessed using bacterial pan-genome analysis program (BPGA), which identifies proteins that are conserved and shared by all diseases to help select vaccine candidates.18 The core proteome was subjected to Cluster Database at High Identity with Tolerance (CD-HIT) analysis to find non-redundant proteins with a cut-off value for sequence identity of 90%.19 The PSORTb website was then used to find the subcellular localization of non-redundant proteins.20,21 Figure 1 depicts the whole methodology’s workflow, and this computational study was conducted in Saudi Arabia in 2022. It is a computational design study. Since no human or animal were involved so ethical approval was not required.

- The workflow of methodology used in current work.
Prioritizing vaccine candidates: Virulent proteins have a crucial role in pathophysiology and thus are suitable candidates for immunization. The Virulent Factor Database (VFDB) categorizes proteins as virulent if they have >30% identity and >100 bit score.22 ProtParam was used to describe physiochemical properties and select pathogen exoproteomes and secretomes for vaccine development.23 Proteins with molecular weight (M.W.) under 110 kDa are more likely to be targets for vaccine. The HMMTOP 2.0 (transmembrane helices and topology prediction server) was used to count transmembrane helices, and proteins containing >1 transmembrane (TM) were given priority for further research.24 Using the VaxiJen server, the antigenicity of the selected proteins was verified, and non-antigenic proteins were eliminated.25 Proteins that were highly antigenic to T-cell receptors and antibodies were discovered to be promising targets.
Prediction of B cell-derived T cell epitopes
Epitopes for these proteins were then predicted using the “immune epitopes database (IEDB)”. The IEDB programme Bepipred Linear Epitope Prediction 2.0 was used to predict linear B-cell epitopes.26 We then mapped T-cell epitopes using selection criteria with a score greater than 0.5. Major histocompatibility complex (MHC) immune receptors are classified into MHC class I and MHC class II; using a percentile score, you can identify epitopes that bind to specific alleles, such as “HLA II (DRB*0101)”.27 High-affinity binding sequences (HABS) were defined as only the epitopes that scored in the bottom percentile. Epitopes with an IC50 (half-maximal inhibitory concentration) value below 100 mm for the primary HLA II allele DRB*0101 were deemed to be potent binders after further optimisation with the MHCPred 2.0 algorithm.28 Several criteria were used to rank epitopes in order of significance. The allergenicity, antigenicity, toxicity, and solubility of a compound were tested using AllerTop, VaxiJen, Toxin pred, and Innovagen, respectively. For the population coverage study, each candidate epitope was evaluated against a global set of alleles.
Multi-epitope vaccine constructs design
Allergen-free, non-toxic, and highly immunogenic epitopes were chosen for the final vaccine formulation. To create a multi-epitope vaccination sequence, we used the IEDB MHC II and MHC I server to predict which B-cells would produce T-cell epitopes. To improve the vaccine’s immunogenicity, the cholera toxin subunit B (CTB) was chosen as an adjuvant and fused to the vector using the EAAAK linker and the GPGPG linker. The CTB sequence was obtained from the Protein Data Bank.29,30
Primary, secondary, and tertiary validation of the vaccine’s model
The physicochemical characteristics of the 3 nascent, hypothetical multi-epitope vaccine constructions were evaluated with the help of the ProtParam ExPASy program. Protein’s secondary structure affects folding. The self-optimized prediction method from alignment (SOPMA) was used to evaluate the model’s secondary structure. It predicted secondary structure with 70% accuracy and counted alpha helices, B-sheet, coil, extended strands, random coils, and beta turns.31,32 Using the Phyre2 server, the construct was modeled, assessed, and validated 3D structure of the vaccine construct. Phyre2 constructed elaborate 3D models by employing cutting-edge distant homology detection strategies. Phyre2 also helps in analyzing the effects of different amino acid variations and predicting where ligands would bind.33 Even though advanced template-based methods are used to describe the 3D structure of an unknown protein, the model structure may still have some errors.34 The lack of similarity between the template and target proteins makes this inevitable. This is due to the fact that structural modifications to the target protein can occur as a result of sequence differences between the template and the target proteins. The Phyre2 server made the 3D structure of the vaccine construct. The GalaxyRefine web server was used to make it better.35 The GalaxyRefine web server does the following: it rebuilds side chains, repacks side chains, and relaxes structures by modeling molecular dynamics.36 Based on the CASP10 evaluation, it has been shown that the best performance has come from the web server method. The ProSA-web (protein structure analysis) server was used to confirm the refined 3D model of the vaccine construct.37 This web server can be used to make new proteins, find mistakes in structures found through experiments, and figure out the total score for a particular structure. If a structure’s numbers fall outside of a certain range, this is called an error.38 This is due to the fact that structural modifications to the target protein can occur as a result of sequence differences between the template and the target proteins. The ElliPro predicted the final 3D model’s conformational B-cell epitopes.39 The software is web-based and aids in the prediction of antibodies against protein antigens based on their sequence. As an ellipsoid, it implements the latest approaches to protein structure analysis.40
Predicting the binding affinity and conformation of the vaccine with the Toll-like receptor (TLR)
Predicting the binding affinity and conformation of a receptor and its ligand is a major goal of bioinformatics, and molecular docking is a powerful technique for doing so. Our study looked into the possibility of binding between the multi- epitope-vaccine model and its receptor. The molecular docking was executed by means of the PatchDock server.41,42 An immunological toll-like receptor 4 (PDB ID: 4G8A) from the Protein Data Bank and a revised 3D model of vaccination were docked together.43,44
Molecular dynamics simulation
The molecular dynamic simulation was employed to analyze the dynamic behavior of the docked complex consisting of the model of the vaccine and TLR4. The atomic-level analysis of the vaccine model demonstrates its flexibility and interactivity through computer-based simulations.45 A simulation of the docked complex was carried out using AMBER20, with a duration of 100 nanoseconds. The ff14SB force field was utilized to document the system topologies. The neutralization of the system was achieved by introducing 3 Na+ ions. Subsequently, the system was placed in a TIP3P water box, with a padding distance of 12. The pre-processing stage was executed, comprising of 7 distinct steps. Initially, the minimization of hydrogen atoms in the system was carried out for 450 cycles, followed by the minimization of the water box for 1000 cycles. The alpha-carbon atoms and non-heavy atoms were minimized for 1000 cycles with a limit of 100 kcal/mol and then for 300 cycles with a limit of 100 kcal/mol.The system underwent a heating process for a duration of 20 picoseconds at a temperature of 300 Kelvin. The utilization of Langevin dynamics was implemented for the purpose of temperature maintenance, with a gamma value of one. The experimental setup involved utilizing a constant-temperature, constant volume (NVT) ensemble to heat the system, while the SHAKE method was employed to limit hydrogen bonding. Subsequently, the system underwent a pre-equilibration phase lasting 100 picoseconds (ps) using a time step of 2 femtoseconds (fs). To achieve pressure equilibration, the isothermal-isobaric (NPT) ensemble was employed, with a restraint of 5 kcal/mol-2 on alpha-carbon atoms. Subsequently, the pressure phase was prolonged by an extra duration of 50 picoseconds, while being restricted to a value of one kilocalorie per mole per 10 picoseconds. Subsequently, the system underwent modifications to accommodate a time scale of one nanosecond. The Berendsen algorithm and an NVT ensemble were utilized to accomplish the production run within a time frame of 100 nanoseconds. The application of the SHAKE method to hydrogen atoms involved the utilisation of an 8.0 cut-off for non-bonded interactions. The assessment of simulation pathways was conducted through the utilisation of AMBER’s CCPTRAJ.
Results
Subtractive proteomic approach shortlisted proteins
With the help of proteins implicated in pathogenesis displayed in Appendix 1, the subtractive proteomics method was employed to select targets for vaccine construction. The proteomes of Sphingobacterium spiritivorum strains was obtained from NCBI. Bacterial pan-genome analysis tool was used to examine core sequence proteins that might be effective vaccine candidates, according to the CD-HIT study, only of the core proteins was discovered to be non-redundant. Non-redundant proteins were next assessed using PSORTb since the sub-cellular localization stage is also critical for proteins to forecast successful vaccine candidates.
Identification of subcellular localization and virulence factors
Prioritization and filtering of screened proteins can contribute to reducing the amount of time, labor, and materials required to generate therapeutic agents and improve the pace of developing the best medication or vaccine against a pathogen. Non-homologous proteins of pathogens could be used as therapeutic and vaccination targets. Additional variables that suggest eligibility of a medication or vaccination target were applied to portray disclosed target proteins. According to the sub-cellular localization of proteins, 25 proteins were found in outermembrane, 9 proteins in the extracellular membrane, and 12 proteins in the periplasmic membrane, these antigenic membrane proteins may be taken into consideration during vaccine design. The hunt for novel VFs is necessary because analysis of virulence factors examined the importance of bacteria in many diseases. According to the VFDB data, these 43 proteins were discovered to be linked to salmonella virulence, demonstrating >30% identity and >100-bit score. These proteins might also be thought of as a crucial target for preventing salmonella pathogenesis. The number of transmembrane helices, on the other hand, was constrained to be either fewer than or equal to one. Due to the challenges in cloning, expressing, and purifying proteins with numerous trans-membranes spanning domains, proteins that were found to have more than one transmembrane helices were excluded. However, none of the chosen membrane proteins had more than one transmembrane helices.
B and T-cell Epitopes selection for vaccine design
Bacterial infection involves humoral immunity. As a result, B-cell epitopes from Sphingobacterium protein candidates were predicted, and antigenicity and surface accessibility were used as selection criteria. Nine peptide fragments overall, with amino acid residues ranging from 25 to 52 mer, were produced. The chosen epitopes underwent MHC-I binding prediction in addition to T cell MHC-II epitope prediction utilizing the IEDB service. These findings led to the identification of 67 epitopes for >core/164/1/Org1 Gene1322 (TonB-dependant receptor (S. spiritivorum) plays role in the transportation of nutrients through the outer membrane), 46 for >core/331/1/Org1 Gene1384 (outer membrane Beta-barrel family protein (S. spiritivorum) helps in membrane biogenesis and signaling), and 27 for >core/1777/1/Org1 Gene2620 (HlyD family secretion protein (Sphingobacterium spiritivorum), it is the part transport pathway and embedded in the inner membrane.46-48 The epitopes with the highest binding affinity to the DRB1*0101* allele were chosen using MHC II pred analysis. This allele is found in all Homo sapiens, and epitopes that bind to it can have powerful immunological effects. The IC50 value was used to calculate this binding capability. The quality of prediction is higher when the IC50 value is low. Predictions revealed a total of 78 epitopes with scores under 100 nm; they should have their antigenicity, allergenicity, toxicity, and solubility evaluated before being accepted as vaccine candidates. We eliminated 26 epitopes because they had non-potent antigenicity, 32 epitopes because they were allergens, and 9 epitopes because they had acceptable solubility.49 Among these epitopes, only 9 epitopes (NAGVNRND, ELKLAKGLK, TLNDKRSSD, ETKRASTDL, GFDPEVGMD, YTFHREDEK, YDDAEIIKV, HADEQRMFS, and KSSLEVSKA) were found to be positive.
Designing multi-epitope-based vaccine construct
Prioritized 9 B-cell derived T cell epitopes were linked together using GPGPG linkers to develop a multi-epitope based vaccine construct. Additionally, CTB adjuvant was attached with an EAAAK linker to the N-terminal end of the first epitope. Cholera toxin B subunit is a potent mucosal adjuvant for the production of mucosal antibody responses and specific immunity.50 The final amino acid sequence of the proposed B-cell derived T-cell based vaccine construct, which comprises a total of 250 amino acids and is a peptide vaccine construct of approximately 26 kDa, is provided below:
MIKLKFGVFFTVLLSSAYAHGTPQNITDLCAEYHNTQIYTLNDKIFSYTESLAGKREMAIITFKNGAIFQVEVPGSQHIDSQKKAIERMKDTLRIAYLTEAKVEKLCVWNNKTPHAIAAISMANEAAAKNAGVNRNDAGPGPGELKLAKGLKGPGPGTLNDKRSSDGPGPGETKRASTDLGPGPGGFDPEVGMDGPGPGYTFHREDEKGPGPGYDDAEIIKVGPGPGHADEQRMFSGPGPGKSSLEVSKA
- Displays the antigenic epitopes that have been filtered and predicted for the 4 vaccine proteins that have been prioritized.
Vaccine safety, antigenicity, solubility and physiochemical properties: According to the findings of the AllerTop online server, the vaccine construct is not allergenic, and the antigenicity scores for VaxiJen and ANTIGENpro were 0.8101 and 0.9144, respectively, indicating that they can effectively elicit a strong immune response. According to ToxinPred and SoluProt prediction results, the construct is non-toxic and water soluble by nature (0.916). The construct’s numerous physiochemical characteristics are listed in Table 2. The molecular weight of the vaccine, which was received through the ExPASy ProtParam server, was around 26 kDalton, and its theoretical (pI) value of 6.6 was estimated, which indicates a somewhat neutral state. The determined aliphatic index was 66.04, the instability index (III) was 27.13, and GRAVY was calculated to be -0.531, all of which indicate that the compound is stable. Additionally, a total of 29 positively charged residues and 30 negatively charged residues, respectively, were calculated. According to data gathered by ProtParam, the vaccine construct’s half-life in Mammalian reticulocytes was calculated to be 30 hours (in vitro), more than 20 hours in yeast (in vivo), and more than 10 hours in E. coli (in vivo). These tests show that vaccine’s structure is very thermostable and hydrophilic, enabling it to interact with blood and water and recognize targets with ease. It is obvious that the components of the developed vaccine match the requirements for vaccine formulation due to their safety, immunogenicity, and stability.51
- Showcases the antigenicity, allergenicity, solubility, and physicochemical properties of the primary sequence of our multi-epitope-based vaccine construct.
Secondary structure prediction and 3D structural modeling, refining, and validation
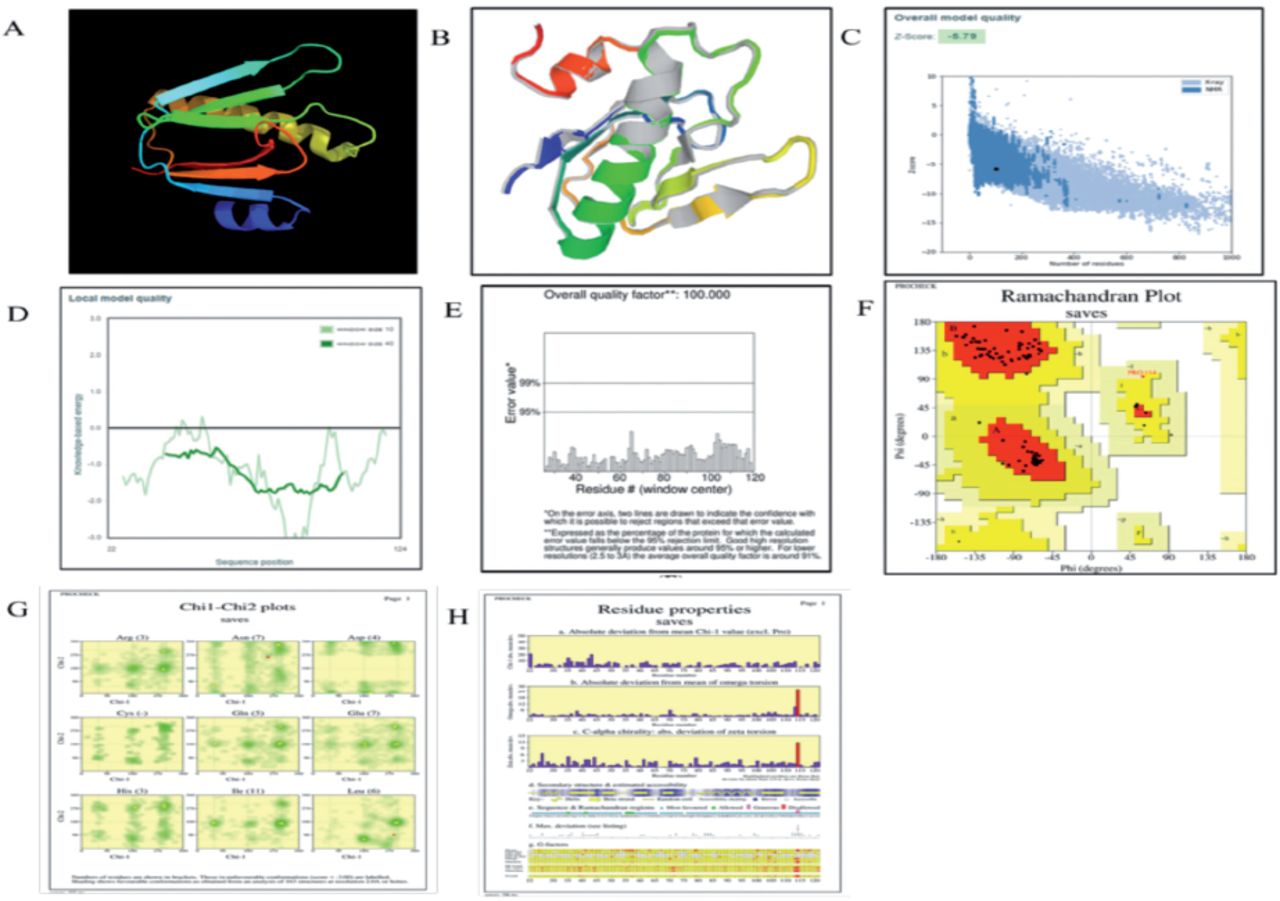
Utilizing the SOPMA web server, the secondary structure of the vaccine was examined.52 The collected data showed that the stable vaccine construct had an alpha helix composition of 28.4%, extended strands of 19.2%, beta turns of 7.6%, and random coils of 48.8%. Additionally, this outcome demonstrated that the vaccine construct’s secondary structure possesses good flexibility, stability, and globular conformation, as illustrated in Appendix 2A-B. The 3D model of the vaccine was predicted through Phyre2. The modeled structure based on template d3chbd was anticipated to be the best template based on a number of 3D structural models of the vaccine that were built, as shown in Figure 2. A single high-scoring template was used to model 103 (or 41%) of the construct’s amino acid residues with 100% confidence. The GalaxyWEB server was also used to refine the 3D vaccine construction model, and it predicted 5 further refined models. Because of its quality scores, which are detailed in Appendix 3 was chosen as the final vaccination model. Predicted B-cell epitopes are shown with a yellow accent, which indicates adequate surface accessibility. The Global distance test high accuracy (GDT-HA) score represents the comparison of 2 protein structures. The GDT-HA score is represented by a number of 0.9806, which is high and denotes a high degree of similarity between the 2 models. The distances between atoms is calculated using the root mean square deviation (RMSD) score; a low RMSD number indicates greater stability. The range of acceptable RMSD scores is 0-1.2, and the model’s RMSD score of 0.367 indicates that protein stability is at a good level. Additionally, the vaccine model has a MolProbity score of 1.365, indicating a decrease in significant mistakes in the 3D model. The model’s clash score was decreased from 24.4 to 6 and represents all undesirable overlapping atoms (evidence of increased stability to high level). A Ramachandran Plot score of greater than 85% represents the surface areas of energetically favorable zones and is considered suitable.

- The present study involves the examination of various aspects related to a vaccine construct, including its 3D crystal structure (A), a refined 3D structure model of the vaccine (B), The refined model’s Z-score is -5.79, which falls within the score range of the residue’s score plot (C), The score plot of Residue by ProSA-web was employed to verify the quality of the local model (D). A ProSA-web validation of the vaccine’s 3D structure (E) An evaluation of the Ramachandran plot for the multi-epitope vaccine construct (F), The number of residues is indicated within brackets. The labeled entities are those that exhibit unfavorable conformations with a score of less than negative three. The utilisation of shading in the analysis of 163 structures at a resolution of 2.0A or higher (G), In conjunction with the properties of residues, reveals a favourable conformation (H).
Validation of model stability
Based on the analysis of the Ramachandran plot, it can be inferred that a significant proportion of the residues (94.7%) in the enhanced protein model are situated within regions that are deemed favourable. The aforementioned score aligns with the GlaxyRefine methodology’s score of 99%. Furthermore, as illustrated in Figure 2, it was found that (F) (5.3%) of residues were located within permissible regions, while none (0%) were situated in prohibited or boundary areas. The PROCHECK server was utilized to determine various structural characteristics of the protein under investigation. Specifically, the server calculated the total number of residues (103), the number of glycine residues (3), the number of proline residues (3), and the number of end residues (2), as illustrated in Figure 2G & H.53
Population coverage and conformational B-Cell epitopes prediction
The epitopes that were chosen underwent population coverage analysis, revealing that they possess the ability to cover 99.78% of the global population, as well as 97.93% of the Chinese population and 97.36% of the Indian population. Appendix 4 illustrates the presentation of various countries and their respective coverage. The conformational B-cell epitope of the 3D-refined model was estimated using the Ellipro server. The server predicted 7 novel conformational B-cell epitopes, which comprised a total of 52 residues. The scores assigned to these epitopes ranged from 0.524 to 0.797. Figure 4 displays comprehensive details of seven epitopes along with a three-dimensional model.
Molecular docking of chimeric protein with TLR4
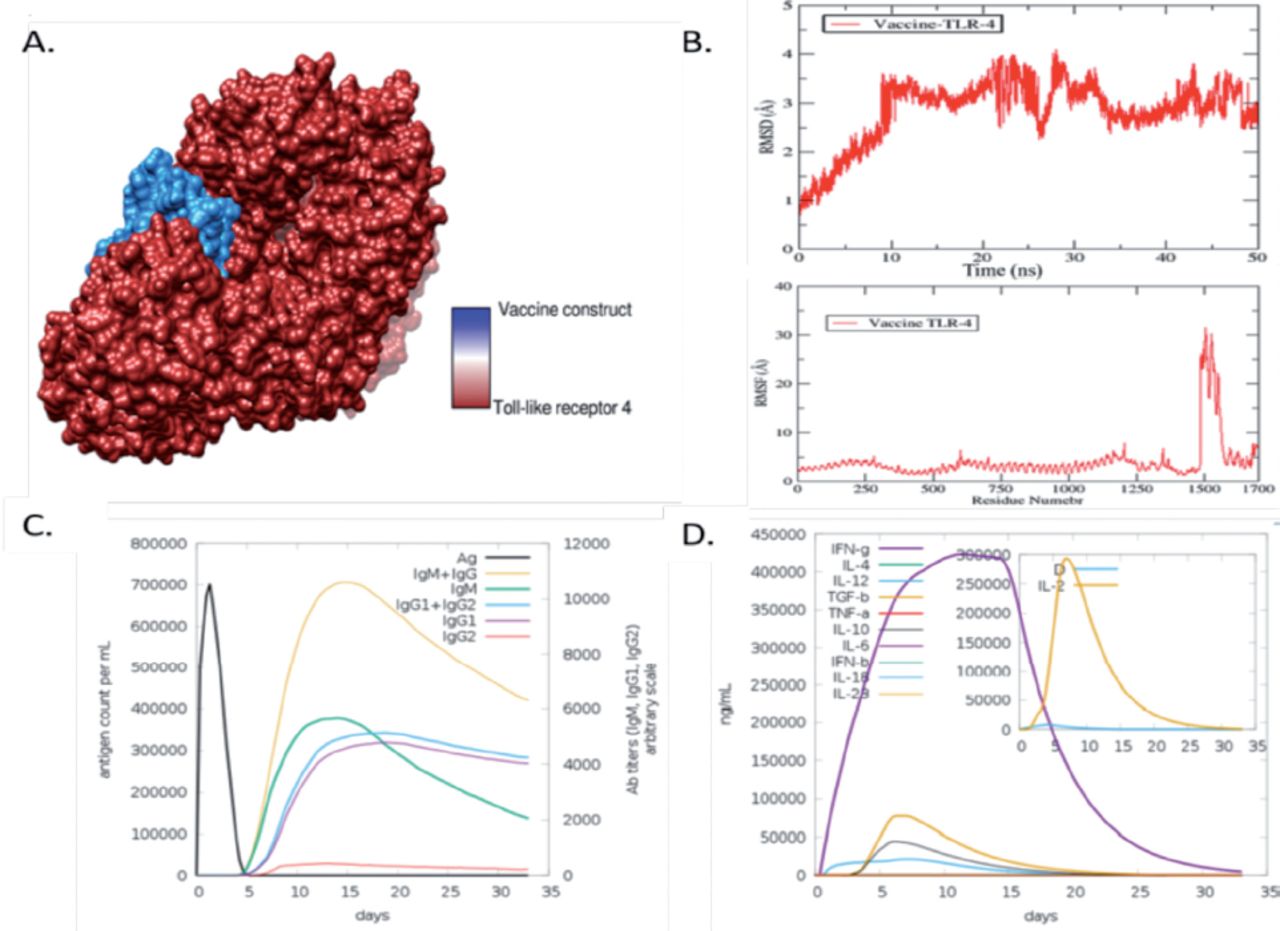
The body can only initiate an effective immunological response upon interaction between an antigenic molecule and a specific immune receptor within the host. Molecular docking analysis was performed to find out the interaction between vaccine and receptors molecules, The TLR4 receptor was subjected to docking with the vaccine designed using PatchDock. A roster of the foremost 10 models was generated according to the PatchDock findings, which evinced significant binding interactions. The selection of these models was predicated upon the conformation of protein surface topology and the degree of electrostatic complementarity. The models obtained were subjected to refinement using the FireDock server. The optimal model was selected from the top 10 models with high binding energy, as determined by PatchDock results (Appendix 5). The fourth solution of the docked complex has been observed to exhibit various energy components, including global energy (-16.61 kJ/mol), attractive van der Waals energy (-43.10 kJ/mol), repulsive van der Waals energy (26.42 kJ/mol), atomic contact energy (13.33 kJ/mol), and hydrogen bond energy (-5.54 kJ/mol), as depicted in Figure 3A.

- Validation and optimization of designed candidate chimeric vaccine. A) The docked complex of the vaccine model and the toll-like receptro (TLR)4 immune receptor. (B) Root Mean square deviation (RMSD) and Root Mean square fluctuation (RMSF) of the vaccine with TLR-4. (C) Antibodies responses against chimeric vaccine predicted by C- immune simulation server. (D) Cytokines and different interferon response induce by chimeric vaccine.
Molecular dynamic simulation
The vaccine-TLR-4 complex was further subjected to molecular dynamic simulation. MD simulation is mainly computer-based simulation in which we observe the dynamic behaviour of the docked molecules. Herein we observed root mean square deviation (RMSD) and root mean square fluctuation of the docked complex of vaccine and TLR-4. In the RMSD analysis of vaccine and TLR-4, we observed a maximum deviation at 30ns at 4.1 (Å) but at the endpoint of simulation, the RMSD showed stability as mentioned in the graph line by red colour in Figure 3B. Furthermore, in root mean square fluctuation (RMSF) analysis, we observed the residual base fluctuation of the vaccine and TLR-4. The RMSF analysis results revealed that the docked complexes showed high fluctuation at the 30 1 (Å) region but showed stability at the endpoint as presented in Figure 3B. The little flexibility is due to the loops present in the docked complex structure.
Binding free energies estimation
Binding free energies were further estimated using MMPB/GBSA analysis. MMPB/GBSA is well-known method for calculation of free binding energies of the docked complex (Vaccine-TLR-4). In this analysis, following different parameter were observed as shown in Table 3.
- Estimation of binding energies.
Codon optimization of chimeric protein and In-silico immune simulation
The Java Codon Adaptation Tool (JCat) was used to optimize the vaccine construct’s codon in E. coli (strain K12) for maximal protein expression. The average GC content for the modified sequence, which was used to determine the improved Codon Adaptation Index (CAI), was found to be 68.8%. Such GC values suggest that the developed vaccine may express itself in a steady manner in the chosen microbial host. The ideal range for a decent GC content is between 30% and 70%. For optimal gene expression, the designed sequence was also included in the E. coli Pet-28a (+) vector. The genetic sequence was cloned into the vector using SnapGene software after inserting a restriction site. Afterward we evaluated the chimeric vaccine construct’s ability to elicit an immunological response in the host body using host immune simulation analyzes. In immune simulation analysis, we observe that the chimeric vaccine has the potency of inducing immune response in the form of IgM and IgG, IgM, of IgM1 and IgG2, and separately IgG1 and IgG2 as presented by different colors peaks in the following (Figure 4C). Furthermore, different interferon level were observer toward chimeric, Maximum IFN-g was observer followed by interleukins 4 (IL-4) and (IL-2) and other tumor growth factor beta (TGF-b) and several other cytokines factors as presented in the following (Figure 3D).
Discussion
In spite of the fact that species of Sphingobacterium are frequently isolated from soil, plants, and water, these bacteria are rarely found at the sites of human infections. Sphingobacterium multivorum and S. spiritivorum were recognised from a limited number of cases that had been previously published. According to Lambiase et al4,5 sputum from individuals with cystic fibrosis was utilized in the process of isolating S. multivorum and S. spiritivorum. It was just recently discovered that an elderly patient was the first human to ever get an infection caused by Sphingobacterium hotanense. In this particular instance, it was suspected that the virus entered the body through scratches that had been made on the right arm by a rooster that had been on the ground nearby. Carbapenems can kill S. spiritivorum.4 The in vitro efficacy of quinolones, trimethoprim-sulfamethoxazole, and ceftazidime is in line with the clinical data that was collected earlier. S. Spiritivorum is a rare organism that can cause cellulitis, and there have only been a handful of documented occurrences of it occurring in the medical literature. Cellulitis was more likely to occur as a result of CHF-related edoema, and ageing and COPD both had the potential to be risk factors for infection. S. spiritivorum is regarded to be a probable cellulitis-causing bacteria, especially in individuals with coexisting risk factors. This is especially true in patients who have had cellulitis in the past.14,54
In this investigation, we utilized the reverse vaccinology approach on the proteome of S. spiritivorum in order to pick the proteins that showed the most promise as potential candidates for a vaccine. Only 2 proteins that were found to be localized to the outer membrane were found to be virulent, antigenic, and non-human homologs. Additionally, these proteins had an appropriate molecular weight and an acceptable number of transmembrane helices (less than 2). It was hypothesised that target proteins would provide B-cells with T-cell epitopes. The top nine epitopes have been selected on the basis of their percentile rank, antigenicity score, allergenicity, toxicity, conservation, and reactivity to a number of different alleles.55,56 This was carried out in order to cover a significant portion of the total population. The epitopes with the highest rankings have been combined using the most effective linkers. The CTB adjuvant was also added in the final design of the multi-epitope vaccination57 in the hopes that it will increase the stimulated immune response and reduce the prevalence of HLA polymorphism in the population.
While preventing the formation of junctions, the GPGPG linker enables the immune system to process and present epitopes that are recognized by HLA-II.58 Additionally, it has been shown that the use of the GPGPG spacer construct can be used to stimulate HTL responses via either polypeptide or DNA vaccination in order to suppress infection.59,60 This can be accomplished by vaccinating the host with either of these. The GPGPG linker was utilized in order to establish connections between CTL epitopes. Additionally, adjuvant was connected together using the EAAAK linker in order to reduce contact with other vaccine domains and to achieve more effective separation.61 As a direct consequence of this, a subunit vaccine consisting of 250 residues was developed.
In addition, codon optimization was performed in order to improve the efficiency of transcription and translation and to allow for high-level expression of the recombinant protein vaccine in E. coli.62 The consequence of increased stability is an increased expression rate. In mammalian reticulocytes, the predicted vaccine had an estimated half-life of thirty hours, while in yeast it had a half-life of more than twenty hours, and in E. coli it had a half-life of more than ten hours, as determined by the analysis of physio-chemical parameters performed by the web server of the ProtParam software.63 In addition, the score for instability was 27.13, which indicates that the protein contained in the subunit vaccine is stable. In addition, the propensity of vaccine solubility upon overexpression in E. coli was calculated to be 0.95 percent, which reveals a decent percentage of solubility in an overexpressed form. According to the findings of the tertiary structure prediction carried out by the Phyre2 server,64 the 3D model contained 28.40% helices and 19.20% sheets. A superior quality 3D model was developed as a result of an in-depth refinement process. The study of all continuous and discontinuous B-cell epitopes revealed that the newly discovered protein surface epitopes were often flexible and could easily interact with antibodies. This was discovered through the examination of all B-cell epitopes.
According to the information that was gathered, the sites 1-26 and 600-617 were where the areas of disorder could be discovered. Intrinsically disordered proteins, often known as IDPs, are proteins that do not have an established or well-structured three-dimensional fold. IDPs feature random coils, (pre-) molten globules, large multi-domain proteins, and flexible linkers. They range from being fully unstructured to being only partially organized in terms of their organization. In many cases, places that are disorganized might be advantageous. When they attach to their respective biological targets, many of the disordered segments fold, while others form flexible linkers, which ultimately results in the formation of macromolecular arrays.65 The proposed construct was then docked with TLR4, and the best docked model was selected and analyzed by MD simulation to determine the stability of the vaccine-protein complex. Lastly, the proposed construct was mutated so that it no longer interacts with TLR4. The results of the MD simulation provide evidence that the proposed interaction between the vaccine and the TLR4 protein would work as expected. The MD simulation improved the way in which the 2 molecules interacted with one another. In silico techniques can be useful instruments for evaluating vaccines, which means that they can be used even before experimental research is carried out.25,66 Therefore, in order to determine the efficacy of the final restricted vaccine, it is necessary to conduct empirical tests on the vaccine that was developed utilizing these immunoinformatics methodologies. In light of this, it is possible to suggest that in vitro synthesis and in vivo experimental studies be carried out in order to analyze the numerous physical and chemical features of the suggested vaccine in order to bring our research on a multi-epitope vaccine construct against S. spiritivorum to a satisfactory conclusion.
In conclusion, S. spiritivorum is an exceptional microorganism that induces cellulitis, with limited documented instances in scholarly literature. The current investigation involves the development of a potent multi-epitope vaccine against S. spiritivorum through the utilisation of various immunoinformatics techniques. Epitopes of B and T cells were forecasted and subsequently narrowed down from the proteins that exhibited potential as vaccine candidates. Linkers and adjuvants were employed to connect the selected epitopes, resulting in the formation of the vaccine construct. The construct was subsequently subjected to an analysis of its physiochemical properties and was modeled and docked with TLR-4. The efficacy of the docked complex was evaluated through molecular dynamics simulations, which demonstrated that the vaccine is generally stable, thereby confirming its effectiveness. The utilisation of immunoinformatics analysis, such as in-silico immune simulation, has demonstrated the efficacy of this vaccine in inducing both humoral and cellular immune responses. This can be attributed to the integration of numerous epitopes derived from a combination of adjuvant and antigens. Hence, it is plausible that the employment of this hybrid vaccine could serve as a therapeutic or prophylactic measure in the management of S. spiritivorum infection sections.
Acknowledgment
This research work was financially supported by the Prince Sattam bin Abdulaziz University (PSAU) under the umbrella of “Deanship of Scientific Research” with project ID 2021/03/19145, therefore, we are extremely grateful to PSAU for their kind and generous support. I would like to acknowledge My Research Essay Help (www.mywritinghelp.com) for the English language editing.
Appendix 1 - Presents the results of the pan-genome analysis. (A) Depicts a phylogenetic tree, while (B) Illustrates the curves of the pan and core genome, and (C) Displays the frequency distribution of gene families within genes. (D) Illustrates the quantity of newly incorporated genes in each respective genome

Appendix 2 - Prediction of models’ quality score by GalaxyWEB
Appendix 3 - Quality scores for selected vaccine model. (A) The secondary structure of the vaccine construct. (B) A secondary structural analysis of the vaccine was conducted, which revealed the fluctuations of its structural atoms within a minimal range, indicating the stability of its structure

Appendix 4 - Docking results of vaccine construct with TLR4 receptor
Appendix 5 -3D model of the 7 predicted conformational B-cell epitopes. The yellow regions are the conformational B-cell epitopes, while the grey regions are the residue remnant. (A) 8 residues with 0.797 score. (B) 7 residues with 0.763 score. (C) 7 residues with 0.663 score. (D) 12 residues with 0.645 score. (E) 8 residues with 0.643 score. (F) 5 residues with 0.551 score. (G) 5 residues with 0.524 score

Appendix 6 - Population coverage analysis predicted by immune database server

Footnotes
Disclosure. This study was funded by the Prince Sattam bin Abdulaziz University, Al-Kharj, Kingdom of Saudi Arabia under the umbrella of “Deanship of Scientific Research” with Project ID 2021/03/19145.
- Received March 16, 2023.
- Accepted April 25, 2023.
- Copyright: © Saudi Medical Journal
This is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Methods
- Results
- Discussion
- Acknowledgment
- Appendix 1 - Presents the results of the pan-genome analysis. (A) Depicts a phylogenetic tree, while (B) Illustrates the curves of the pan and core genome, and (C) Displays the frequency distribution of gene families within genes. (D) Illustrates the quantity of newly incorporated genes in each respective genome
- Appendix 2 - Prediction of models’ quality score by GalaxyWEB
- Appendix 3 - Quality scores for selected vaccine model. (A) The secondary structure of the vaccine construct. (B) A secondary structural analysis of the vaccine was conducted, which revealed the fluctuations of its structural atoms within a minimal range, indicating the stability of its structure
- Appendix 4 - Docking results of vaccine construct with TLR4 receptor
- Appendix 5 -3D model of the 7 predicted conformational B-cell epitopes. The yellow regions are the conformational B-cell epitopes, while the grey regions are the residue remnant. (A) 8 residues with 0.797 score. (B) 7 residues with 0.763 score. (C) 7 residues with 0.663 score. (D) 12 residues with 0.645 score. (E) 8 residues with 0.643 score. (F) 5 residues with 0.551 score. (G) 5 residues with 0.524 score
- Appendix 6 - Population coverage analysis predicted by immune database server
- Footnotes
- References
- Figures & Data
- eLetters
- References
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.