


Abstract
Central alveolar hypoventilation is rarely encountered. This case report describes a young woman who was recently diagnosed with hypertension and ischemic heart disease. She presented to the emergency room with hypercapnic respiratory failure, for which she was mechanically ventilated. This was preceded by an acute upper respiratory tract infection. She was initially suspected to have Guillain-Barré syndrome, but further investigations ruled out neuromuscular or autoimmune disorders. Sleep-related hypoventilation was suspected after she experienced recurrent apneas at night that resulted in re-intubation. Polysomnographic studies confirmed episodes of central apnea and hypopnea during sleep, with significant carbon dioxide retention and oxygen desaturations. She required nocturnal ventilation via a tracheostomy tube until a diaphragmatic pacer could be placed. Using bi-level positive airway pressure and average volume-assured pressure support together with the diaphragmatic pacer, adequate ventilation during sleep was achieved.
Sleep related hypoventilation is characterized by abnormal respiration and high carbon dioxide (PCO2) during sleep. It is associated with absent or greatly diminished ventilatory response to hypercapnia. This occurs mainly in patients with central nervous system pathology, neuromuscular diseases, and in obesity hypoventilation syndrome.1,2 Rarely, central hypoventilation can be due to congenital disorders of the autonomic nervous system, that control breathing, such as the congenital central hypoventilation syndrome (CCHS), where there is loss of the normal autonomic control of breathing due to a functional derangement of the center of breathing in the brainstem.3,4 The resultant derangement in gas exchange can progress to hypercapnic respiratory failure under stressful conditions.1,2-4 The objective of this case report is to describe a rare case of central alveolar hypoventilation in an adult female with history of unexplained hypertensive and cardial ischemia.
Case Report
Patient information
A 23-year-old Syrian female college graduate was diagnosed with hypertension 6 months prior to her acute presentation. Secondary causes of hypertension were ruled out. Two months later, she presented with recurrent attacks of chest pain; during one attack, electrocardiographic changes were suggestive of non-ST elevation myocardial infarction and laboratory tests were positive for troponin I. Coronary angiography was carried out and revealed normal coronary arteries. At presentation to the emergency department, she was found to have bilateral lower limb pain and weakness in the legs. She reported an upper respiratory tract infection that had started a few days earlier. Chest x-ray showed bilateral basal atelectasis. Shortly after admission, she progressed to hypercapnic respiratory failure (PaCO2 87 mm Hg) and she was intubated and put on mechanical ventilation. (Table 1).
Timeline of relevant medical history and interventions.
Clinical findings
On clinical examination, she was fully conscious and communicating. Her pupils were 3 mm and reactive to light bilaterally, with no ptosis. Vital signs showed a respiratory rate of 18 breaths/min, blood pressure of 140/95 mm Hg, pulse of 112 beats/min, and she was afebrile. Her body mass index (BMI) was 21 kg/m2, and she had no lower limb edema. Cranial nerve examination revealed no abnormalities. Motor power was 5/5 in the upper limbs and 4/5 in the distal lower limbs. No sensory deficits were elicited. Plantar reflexes were down going bilaterally.
Diagnostic assessment
On laboratory examination, complete blood count, renal function, thyroid function, and muscle enzymes were all normal. The initial differential diagnoses included Guillain-Barré syndrome (GBS), myasthenia gravis (MG), autoimmune diseases, muscular dystrophy, and paraneoplastic syndrome. Cerebrospinal fluid examination showed normal protein and no inflammatory cells. Magnetic resonance imaging (MRI) of the brain and the whole spine were normal. Magnetic resonance angiography (MRA) of the brain showed that all major arteries were normal. Computed tomography of the chest, abdomen, and pelvis was unremarkable. Repeat MRI/MRA of the brain with contrast was performed after 2 weeks and was unremarkable (Table 1).
On nerve conduction study (NCS), compound muscle action potentials and sensory nerve action potentials were all normal. Repetitive nerve stimulation test of the right abductor pollicis brevis muscle was normal. There was no decrement or increment to suggest a neuromuscular junction disorder. Nerve conduction study of both phrenic nerves was repeated twice (3 weeks apart) and revealed normal distal latencies and amplitudes. Needle electromyography (EMG) studies of the left diaphragm deltoid, biceps, first dorsal interosseous, and tibialis anterior muscles were normal. Serology for autoimmune vasculitis and beta-2 glycoprotein antibodies was negative. Work-up for MG was negative. Voltage-gated calcium channel antibodies were also normal. Echocardiography showed normal ejection fraction (>55%) and no thrombus, but mild concentric left ventricular hypertrophy. Muscle biopsy from the tibialis anterior muscle was normal and no inflammation was seen.
Therapeutic intervention
Upon admission to the intensive care unit (ICU), she received 2 doses of intravenous immunoglobulins, and was started on steroids and azathioprine for possible GBS or autoimmune vasculitis. However, these medications were stopped after one week in view of the negative investigation results and quick normalization of muscle power. She was extubated twice but had to be re-intubated at night due to apnea and acute rise in PCO2 followed by oxygen desaturation during sleep. She eventually underwent tracheostomy after the second re-intubation.
Post-tracheostomy sleep studies were conducted in the ICU. The results were consistent with central hypoventilation and lack of ventilatory and arousal response to hypercapnia during sleep (Table 2). Despite the rise in PCO2 after disconnecting the ventilator for 45 min, no respiratory effort was recorded (Figure 1). Genetic testing for PHOX2B, which is diagnostic for congenital central hypoventilation syndrome (CCHS), was negative.
Arterial blood gases during the first sleep study in ICU.
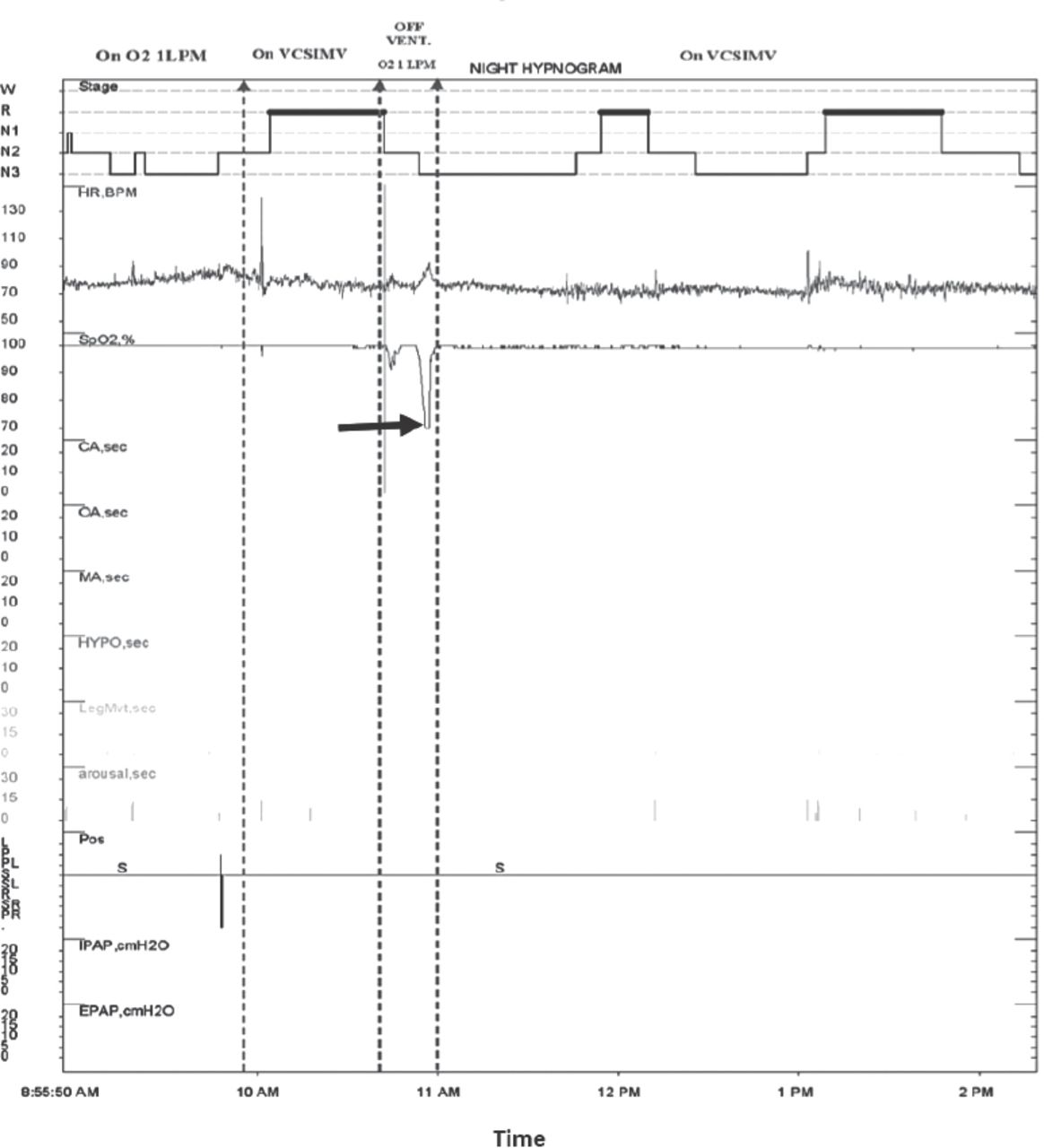
Sleep study. Epoch displaying desaturation off mechanical ventilation while on tracheostomy mask (arrow). Stage - sleep stage, HR - heart rate, BPM - beat per minutes, SPO2 - oxygen saturation, CA - central apnea, OA - obstructive apnea, MA - mixed apnea, HYP - hypopnea, Desat - desaturation durati, Pos - position, S - supine, IPAP - inspiratory, positive airway pressure, EPAP - expiratory positive airway pressure
Central stimulants were tried without success. During sleep, she was mechanically ventilated (volume control-synchronized intermittent mandatory ventilation) but when she was awake, she was fully mobile on room air. After 2 weeks of nocturnal ventilation, diaphragmatic pacer, NeuRx®, Synapse Biomedical Inc. Oberlin, Ohio, US, was placed via a thoracoscopic approach (set at pulse frequency 18/min, inspiration interval 1.3 s, pulse ramp 10). The tracheostomy was closed, and all antihypertensive medications were stopped after the nocturnal hypoventilation was eliminated (Table 1).
Follow up and outcome
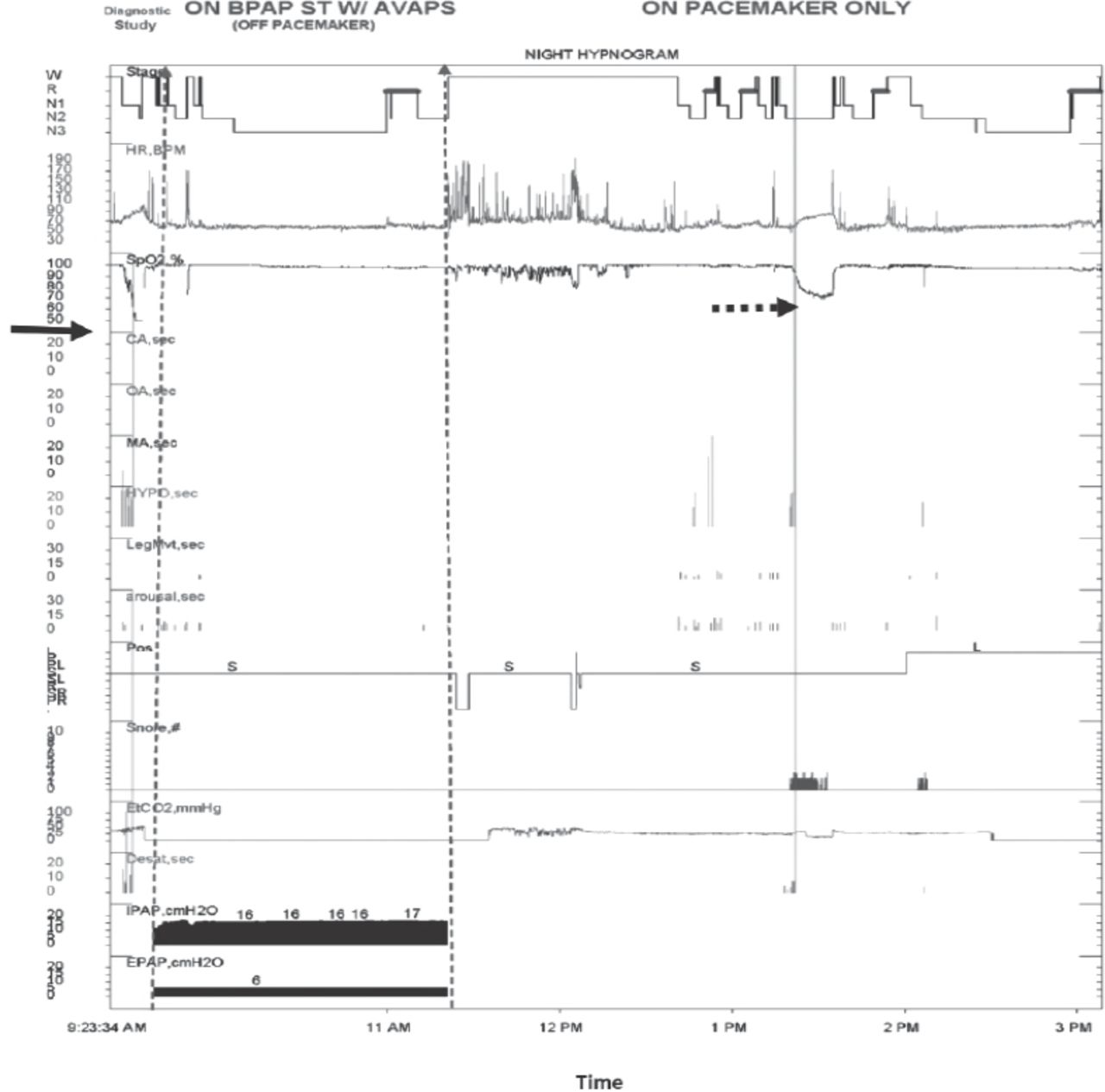
Following repeat sleep studies, she was commenced on bi-level positive airway pressure (BiPAP) at 16/6 with spontaneous/timed (S/T) mode (timed inspiration 1.5 s, back-up rate 12) along with average volume-assured pressure support (AVAPS) and discharged home. Seven months later, a 3-part split sleep study was conducted: 1) a diagnostic study with no intervention (off BiPAP and off pacemaker); 2) on BiPAP ST-mode with AVAPS as in her home settings; and 3) on pacemaker only (18/min, inspiration interval 1.3 s). During the diagnostic section, which lasted 6 min, she had prolonged hypoventilation events that resulted in significant desaturation down to 20%. However, on BiPAP S/T-mode with AVAPS, no respiratory events were detected. On pacemaker only, central hypoventilation was observed with desaturation down to 67% (Figure 2). This study indicated the persistence of central hypoventilation and the inadequate response to diaphragmatic pacer alone but complete elimination of all central hypopneas with BiPAP S/T-mode with AVAPS.

Sleep study epoch displaying sleep realted hypoventilation during diagnostic part (solid arrow), which was eliminated by BPAP ST/T mode and significant desaturation on pacemaker alone (broken arrow). Stage - sleep stage, HR - heart rate, BPM - beat per minutes, SPO2 - oxygen saturation, CA - central apnea, OA - obstructive apnea, MA - mixed apnea, HYP - hypopnea, Desat - desaturation durati, Pos - position, S - supine, L - left, IPAP - inspiratory, positive airway pressure, EPAP - expiratory positive airway pressure
Discussion
Patient perspective
We describe a case of central alveolar hypoventilation in a young adult female from the Middle Eastern region. The diagnosis of this patient was challenging. The initial presentation with lower limb pain and weakness suggested GBS; however, NCS and central spinal fluid analysis were normal. Similarly, other central nervous system lesions and autoimmune disorders were ruled out. The observation of recurrent episodes of central apnea, hypopnea during sleep, CO2 retention, and oxygen desaturation suggested a diagnosis of sleep-related hypoventilation.1,2 Sleep studies confirmed the presence of central hypoventilation. Common causes of sleep-related hypoventilation such as medications and chronic medical conditions were ruled out. Obesity hypoventilation syndrome was not entertained as the patient had normal BMI and daytime PCO2. The diagnosis of CCHS was strongly considered.3 Although classically described in neonatal period, late onset congenital CCHS have been rarely reported in adults.4
Respiratory tract infections, medications or general anesthesia can precipitate or precede the clinical presentation of CCHS.4,5 This patient gave history of upper respiratory tract infection (URTI) 5 days preceding the clinical presentation. Similarly, Butin et al6 described a 9-year-old patient who was diagnosed CCSH following hypercapnic respiratory failure that was caused by mycoplasma pneumonia. A case report from Oman5 described a 6-year old child who was diagnosed as late onset CCHS after witnessed hypoventilation during sleep following general anesthesia for dental surgery. Although the clinical picture and sleep studies were consistent with this syndrome, genetic testing for PHOX2B gene mutation was negative. It is possible that this patient has other rare genetic mutations that require more detailed DNA sequencing. Alternatively, if DNA sequencing is negative, the diagnosis of idiopathic central alveolar hypoventilation can be made as per the International Classification of Sleep Disorders.1 The recent history of hypertension in this patient could be related to the effect of hypoxemia and hypercapnia on sympathetic stimulation, cardiac contractility, and heart rate.7 This normalized after correction of apnea.
The management of patients with central alveolar hypoventilation is directed at providing assisted ventilation during sleep.2 Electrical stimulation of the diaphragm can be used to reduce dependence on mechanical ventilation.8 Similar to our patient, a recent report described successful decannulation of tracheostomy among several patients with central hypoventilation.9 In the present patient, EMG of the diaphragm and NCS of the phrenic nerve were undertaken before pacing, and were normal. However, despite initial response to the pacer that helped to decannulate the patient to BiPAP, the follow-up sleep studies showed that pacer alone was inadequate. The use of BiPAP S/T-mode with AVAPS back-up resulted in complete resolution of nocturnal hypoventilation and maintained adequate tidal volume during sleep. This BiPAP mode delivers fixed inspiratory and expiratory positive airway pressure. Average volume-assured pressure support adjusts the inspiratory pressure support according to target tidal volume,9 and has been used successfully in patients with chronic obstructive pulmonary disease and obesity hypoventilation syndrome.10
In conclusion, this is a rare case of central alveolar hypoventilation in a young woman that was precipitated by upper respiratory tract infection. Adequate nocturnal ventilation was achieved with diaphragmatic pacing in conjunction with BiPAP and AVAPS. Central alveolar hypoventilation needs to be considered in young patients presenting with unexplained hypertension and cardiac symptoms and hypercapnic respiratory failure. A multidisciplinary approach and early referral to a sleep center is needed for proper diagnosis and management of this rare but serious condition.
Withdrawal policy
By submission, the author grants the journal right of first publication. Therefore, the journal discourages unethical withdrawal of manuscript from the publication process after peer review. The corresponding author should send a formal request signed by all co-authors stating the reason for withdrawing the manuscript. Withdrawal of manuscript is only considered valid when the editor accepts, or approves the reason to withdraw the manuscript from publication. Subsequently, the author must receive a confirmation from the editorial office. Only at that stage, authors are free to submit the manuscript elsewhere.
No response from the authors to all journal communication after review and acceptance is also considered unethical withdrawal. Withdrawn manuscripts noted to have already been submitted or published in another journal will be subjected to sanctions in accordance with the journal policy. The journal will take disciplinary measures for unacceptable withdrawal of manuscripts. An embargo of 5 years will be enforced for the author and their co-authors, and their institute will be notified of this action.
Acknowledgment
We would like to thank Prof. Ahmed S. Bahammam for his valuable contribution to the case study and analysis of the sleep study results.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received December 4, 2017.
- Accepted January 10, 2018.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.




{kind=link}
{kind=link}