


Abstract
Objectives: To identify the underlying gene mutation in a large consanguineous Pakistani family.
Methods: This is an observational descriptive study carried out at the Department of Biochemistry, Shifa International Hospital, Quaid-i-Azam University, and Atta-ur-Rahman School of Applied Biosciences, National University of Sciences and Technology, Islamabad, Pakistan from 2013-2016. Genomic DNA of all recruited family members was extracted and the Trusight one sequencing panel was used to assess genes associated with a neuro-muscular phenotype. Comparative modeling of mutated and wild-type protein was carried out by PyMOL tool.
Results: Clinical investigations of an affected individual showed typical features of Miyoshi myopathy (MM) like elevated serum creatine kinase (CK) levels, distal muscle weakness, myopathic changes in electromyography (EMG) and muscle histopathology. Sequencing with the Ilumina Trusight one sequencing panel revealed a novel 22 nucleotide duplication (CTTCAACTTGTTTGACTCTCCT) in the DYSF gene (NM_001130987.1_c.897-918dup; p.Gly307Leufs5X), which results in a truncating frameshift mutation and perfectly segregated with the disease in this family. Protein modeling studies suggested a disruption in spatial configuration of the putative mutant protein.
Conclusion: A novel duplication of 22 bases (c.897_918dup; p.Gly307Leufs5X) in the DYSF gene was identified in a family suffering from Miyoshi myopathy. Protein homology analysis proposes a disruptive impact of this mutation on protein function.
Dysferlinopathies are a genetically originated group of the heterogeneous disorders which include autosomal recessive muscular dystrophies.1,2 These conditions are designated by muscular weakness and muscular atrophy, while in a few cases symptoms were restricted to distal muscles. Miyoshi myopathy (MM-MIM #254130) is a recessively inherited distal type of muscular dystrophy that presents at early adulthood. It clinically resembles other myopathies which involves the distal musculature, especially the muscles of lower legs.3 The mutations in the DYSF gene are a major cause of this disease.4 It differs from LGMD2B, in which the proximal part of muscles are affected although other characteristics such as age of onset, higher creatine kinase (CK) levels, progressive disease pattern, and dysmorphology of muscles have been found as well.5
DYSF encodes a 237-kDa dysferlin sarcolemmal trans-membrane protein having membrane repair function through regulation of vesicle fusion with the sarcolemma and to a smaller extent with secretory vesicles of the Golgi network.6 It is also involved in membrane fusion, angiogenesis, myogenesis, and microtubule function.7 The DYSF gene (NM_001130987) comprises of 6.6 kb nucleotide sequence with 56 coding exons, encoding 2119 amino acids protein. DYSF mutations are dispersed throughout the gene, and more than 500 pathogenic nucleotide variants are documented in Leiden Muscular Dystrophy Database for dysferlinopathy, showing different effects on protein function.8 Among the 122 reported sequence variations in the DYSF, 10 duplications were reported in several families and sporadic cases so far causing Miyoshi myopathy phenotype.4,7-12
In this study, we report a consanguineous family of Pakistani origin with a phenotype consistent with Miyoshi myopathy. We identified a novel homozygous duplication/frameshift mutation of DYSF gene (c.897_918dup; p.Gly307Leufs5X) in affected siblings of the family.
Methods
Prior to start of the study approval was granted from Advance Studies & Research Board (AS&RB) of National University of Sciences and Technology, Islamabad, Pakistan. This was an observational descriptive study. This was carried out at Department of Biochemistry, Quaid-i-Azam University, Islamabad and National University of Sciences and Technology, Islamabad, Pakistan during 2013-2016. The study was carried out according to the Helsinki declaration with amendments made at 64th World Medical Association, General Assembly, Fortaleza Brazil, October, 2013.
Inclusion criteria were family having multiple affected individuals with specific phenotype of Miyoshi myopathy. Exclusion criteria were families with a single affected member were excluded from this study.
Recruitment of patients/family
A consanguineous Pakistani family with 4 affected individuals was presented to Division of Neurology, Shifa International Hospital Islamabad, Pakistan, and was clinically diagnosed as MM. The pedigree was consistent with autosomal recessive inheritance (Figure 1A) with 4 affected individuals (V-3, V-4, V-5, and V-7). Blood samples were available from 4 affected and 6 unaffected family members. Informed consent was taken from the recruited family individuals of the study after permission from the Institutional Review Board of Shifa International Hospital, Quaid-i-Azam University and National University of Sciences and Technology (NUST), Islamabad, Pakistan.
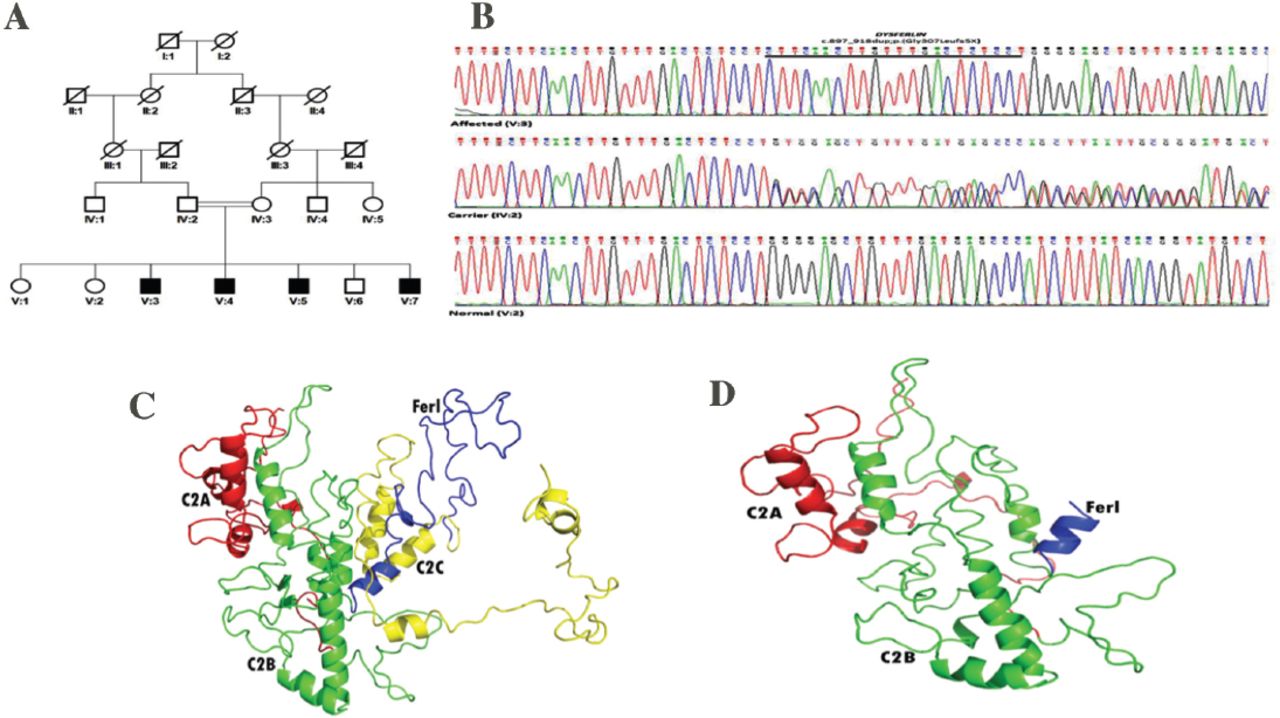
Mutation analysis and homology modeling of DYSF. A) Pedigree of multi-generation Pakistani family showing co-segregation of a novel duplication (c.897_918dupCTTCAACTTGTTTGACTCTCCT) with the disease and B) sequence electropherograms for a wild-type (V:2), heterozygous (IV:2) and homozygous individual (V:3) with 22 nucleotide duplication in DYSF. Comparative homology modeling proposed distinct alterations in dysferlin protein while comparing the C) wild-type with D) mutant type.
Trusight one sequencing panel and Sanger sequencing
Extraction of genomic DNA was carried out from the Ethylenediaminetetraacetic acid (EDTA) whole blood using the QIAmp DNA blood Mini-Kit (QIAGEN, USA).Targeted sequencing was carried out in one affected individual of Miyoshi myopathy family by using trusight one sequencing panel technique for genes of neurological disorders (especially of muscular dystrophy phenotype). The sequencing coverage area with targeted genes (<50 genes) panel was <15X coverage to attain adequate methodological sensitivity.13 Sanger sequencing was performed by using CEQ8000 Genetic analyzer (Beckman Coulter, USA) for exon 9 of DYSF gene according to the protocols described previously.14
Comparative protein modeling
To model the three-dimensional structure of the variant protein, analysis for dysferlin protein was performed with BLASTp software by searching sequences in protein database (PDB). No significant hits found. Therefore, an alternative procedure for modeling was carried out with an Iterative Threading ASSEmbly Refinement (I-TASSER) tool.15 The predicted protein 3-D structure was visualized by PyMOL (http://pymol.org/).
Results
Clinical description of Patient 1 (V:3)
A 24 years old man presented with difficulty in rising from the floor, onset at 17-18 years age. Three of his brothers were also affected and his parents were first cousin (Figure 1A). He had a waddling gait and the Gower sign was positive (Table 1). He had progressive wasting and weakness of deltoid, biceps, and triceps, and tapering of the half of forearm quadriceps. Investigations revealed markedly elevated serum creatine kinase and lactate dehydrogenase (13,100 U/L and 1,364 U/L respectively). Muscular power using Medical Research Council (MRC) grade was 4+/5 in the deltoid muscles bilaterally, biceps 4/5, grip 4+/5, hip flexors were 3-/5 and knee extensors 4-/5 bilaterally. Deep tendon reflexes (DTR’s) were 1+, symmetrical in upper limbs and absent in lower limbs while planter reflexes were bilateral flexor. Electromyography (EMG) showed myopathic changes. Calf muscle biopsy done at outside facility showed muscular dystrophy. The clinical, laboratory and histological features were consistent with Miyoshi myopathy. Figure 2 illustrates the muscle wasting in this affected individual.
Clinical detail of affected individuals of Miyoshi myopathy family.

Images of affected individual (V:3) of Miyoshi myopathy family showing atrophy of A) proximal muscles, B) forearm muscles, and C) intrinsic muscles of the hand.
Patient 2 (V:4)
A 26-year-old male presenting with a history of progressive gait difficulty since 18 years of age, difficulty in climbing stairs, and difficulty in arising from the floor. At age of 20 years, he also developed bilateral mild weakness of his grip. His physical and clinical evaluations are described in Table 1.
Patient 3 (V:5)
A 31-year-old male presented with progressive muscle wasting of distal forearms since the age of 17-18 years. Features are explained in Table 1. Power in the upper limb was MRC grade 2/5 proximally and 4/5 distally. Hip flexors were MRC grade 2/5 bilaterally, quadriceps muscle were MRC grade 2/5 and dorsi-flexor of the feet 3+/5 bilaterally. Deep tendon reflexes were all absent and the plantar responses were bilateral flexor (Table 1).
Patient 4 (V:7)
A 35-year-old male who was wheel chair bound and presented with progressive weakness of his legs since 17-18 years of age. His physical features and clinical investigations are mentioned in Table 1. On physical examination, he was alert and oriented with normal speech. Muscle power in upper limbs was MRC grade 1/5 bilaterally, while grip strength was 0/5 bilaterally. His lower limb power was 1/5 proximally and 1/5 distally. Deep tendon reflexes were all absent and plantar responses were bilateral flexor.
Mutation detection and comparative analysis. The potential pathogenic candidate variants from the panel sequencing data were assessed by Mutation Taster for pathogenicity prediction. We focused on a DYSF variant as it was the most suitable candidate gene. To confirm the duplication identified by panel sequencing, unique primers of exon 9 of DYSF were designed and PCR amplified. Purified products were Sanger sequenced using dye-terminator chemistry and electrophoresed on CEQ8000 Genetic analyzer (Beckman Coulter, USA). A novel frameshift variant [(NM_001130987.1; DYSF_v001): c.897_918dupCTTCAACTTGTTTGACTCTCCT; p.(Gly307Leufs5X)] was confirmed and co-segregated with the phenotype in this family (Figure 1B). This variant was absent in dbSNP and EXAC Browser (http://exac.broadinstitute.org/) databes. It was also absent in 50 chromosmes from Pakistani ancestry. Protein homology modeling was performed and analyzed for this truncated protein in comparison to wild structure of protein (Figure 1C); it was observed a significant change of spatial configuration in mutant type (Figure 1D) resulting in probable deleterious functioning.
Discussion
In this study, we identified a novel frameshift variation in DYSF namely a duplication of a 22 nucleotides sequence, in all affected individuals of a family suffering from Miyoshi myopathy. All affected individuals presented with wasting and weakness of distal half of the muscles from forearms and legs. They also had weakness of grip, difficulty in walking, climbing stairs and raising their arms. Patients (V-3 and V-4) had clinical and electrophysiological features of MM. One affected individual, (V:3) had dystrophic features on muscle histopathology. Candidate gene sequencing revealed a novel 22 base pair duplication mutation (c.897_918dup; p.Gly307LeufsX5) in DYSF gene. Protein modeling results predicted the significant alteration in the spatial configuration of mutant type of dysferlin protein.
Miyoshi myopathy is clinically heterogeneous with similar features such as other dysferlinopathies.2,20 In MM, the weakness progresses to involve the hamstring muscles, and later, the hip and pelvic girdle. The extensor muscles of the forearms may also become weak and atrophic, but the brachio-radialis and hand intrinsics are typically spared. The rate of progression can be quite variable with some individuals progressing rapidly over a few years to being non-ambulatory, however one-third of affected cases were found wheelchair bound after 10 years of onset.21 Katz et al22 described patients with a Miyoshi phenotype that did not have a primary dysferlinopathy. These patients differed from those with dysferlinopathy by their later age of onset (usually after 30 years of age) and discreetly raised serum CK level.22 In the present study, the clinical phenotype of Miyoshi myopathy is consistent with already reported phenotypes.4,8
The causative mutations in DYSF (MIM# 603009) are known cause of muscular dystrophies like Miyoshi myopathy and LGMD2B.4 Due to overlapping phenotypes with other forms of muscular dystrophies, muscle biopsy and genetic analysis of DYSF are considered to be the gold standard for diagnosis of Mioshi myopathy.7 Panel diagnostics through TruSight one sequencing panel (www.illumina.com) is suitable technique to identify pathogenic variants in muscular dystrophies, especially in families with 1 affected individual; where homozygosity mapping is not possible. However, in some muscular dystrophies, hybridization may be the best technique.
Generally, the pathogenic mechanism of diseases stated that higher expression of dysferlin is observed at the level of injury in membrane. Some comprehensive studies about dysferlin null mice and micro-injury of myofibers have showed that the membrane repair mechanism is significantly impaired due to DYSF lacking muscle activity.23,24
In dysferlinopathies, approximately 500 diverse sequence variations, including deletion mutations and other non-pathogenic polymorphisms, are described in Leiden Muscular Dystrophy database [Leiden Muscular Dystrophy pages (http://www.umd.be/DYSF/)]. Out of total DYSF mutations, about 122 mutations are associated with Miyoshi myopathy [HGMD Professional 2015.2]. Among these mutations, 10 mutations are frameshift mutations due to small insertion or duplication of nucleotide sequences (Table 2). In present study, we have identified a 22 base pair duplication mutation (CTTCAACTTGTTTGACTCTCCT) which results in the shifting of reading frame up to five amino acids and truncating protein due to premature stop codon creation (p.Gly307Leufs5X). This is the largest duplication reported till present. According to published data, this is the first report of clinical and molecular investigations in a large family Pakistani family suffering from Miyoshi myopathy. Although, the highly consanguineous Pakistani population has shown hundreds of autosomal recessive pedigrees of different disorders (NCBI), less attention is given to complex neuro-muscular disorders like Miyoshi myopathy mainly due to lack of clinical facilities.
Previously reported insertion/duplication mutations in DYSF gene associated with Miyoshi myopathy.
In conclusion, clinical and molecular investigations in a multi-generation Pakistani pedigree showing the symptoms of Miyoshi myopathy revealed a novel duplication of 22 bases (c.897_918dup; p.Gly307Leufs5X) in DYSF gene. In silico studies propose a disruptive impact of this mutation on protein function.
Acknowledgment
We are grateful to the family members who participated in this research study. We are thankful to Dr. Muhammad F. Bhatti (PhD, Imperial College London, UK) for critically reviewing language of the manuscript.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received March 15, 2017.
- Accepted July 31, 2017.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial License (CC BY-NC), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.




{kind=link}
{kind=link}