


Abstract
Objectives: To use independent transcriptomics data sets of cancer patients with prognostic information from public repositories to validate the relevance of our previously described chronic lymphocytic leukemia (CLL)-related proteins at the level of transcription (mRNA) to the prognosis of CLL.
Methods: This is a validation study that was conducted at Majmaah University, Kingdom of Saudi Arabia between January-2017 and July-2018. Two independent data sets of CLL transcriptomics from Gene Expression Omnibus (GEO) with time-to-first treatment (TTFT) data (GSE39671; 130 patients) and information about overall survival (OS) (GSE22762; 107 patients) were used for the validation analyses. To further investigate the relatedness of a transcript of interest to other neoplasms, 6 independent data sets of cancer transcriptomics with prognostic information (1865 patients) from the cancer genomics atlas (TCGA) were used. Pathway-enrichment analyses were conducted using Reactome; and correlation analyses of gene expression were performed using Pearson score.
Results: Nine of the CLL-related proteins exhibited transcript expression that predicted TTFT and 7 of the CLL-related proteins showed mRNA levels that predicted OS in CLL patients (p≤0.05). Of these transcripts, 8 were different types of heterogeneous nuclear ribonucleoproteins (HNRNPs); and 2 (HNRNPUL2 and HIST1C1H) retained prognostic significance in the 2 independent data sets. Furthermore, genes that enriched CLL-related pathways (p≤0.05; false discovery rate [FDR] ≤0.05) were found to correlate with the expression of HNRNPUL2 (Pearson score: ≥0.50; p<0.00001). Finally, increased expression of HNRNPUL2 was indicative of poor prognosis of various types of cancer other than CLL (p<0.05).
Conclusion: The cognate transcripts of 14 of our CLL-related proteins significantly predicted CLL prognosis.
Chronic lymphocytic leukemia (CLL) is a malignant disease that affects B-cells and results in the accumulation of leukemic cells in the peripheral blood and lymphoid tissues.1 Chronic lymphocytic leukemia is an adult disease that predominantly affects males; the male-to-female incidence ratio of the disease is 2:1.2 Advanced treatment modalities of CLL enable significant improvements in overall survival and life quality of afflicted patients.3 However, the disease is still incurable and life-threatening for many patients.4 Chronic lymphocytic leukemia is a heterogeneous disease with a variable clinical course.5 Some patients have a stable form of CLL with no or late need for treatment and long overall survival. However, others exhibit an aggressive form of the disease with an early need for therapy and short overall survival. Various molecular prognostic markers have been well-established and commonly applied to predict the clinical outcomes of CLL.5 Unmutated immune globulin heavy variable genes (IGVH) indicate high-risk CLL, and mutated IGVH are associated with low-risk CLL.6 In addition, elevated expression of CD38 and tyrosine-protein kinase 70 (ZAP-70) is a characteristic of an aggressive form of CLL.7,8 Chromosomal aberrations such as deletions in q11 and p17 are informative markers of poor prognosis of CLL; a deletion in 13q indicates a favorable prognosis of the disease.9 Although these prognostic markers offer significant aid in predicting the clinical course of CLL, the prognostication of the disease remains challenging.10 Proteomic approaches offer a valuable opportunity for the discovery of disease- related proteins.11 In our previous work, we applied qualitative and quantitative proteomic approaches to explore the proteome of CLL samples from 12 patients with different prognoses.12,13 Our findings described 63 candidates as CLL-related proteins. The relevance of 4 of these proteins to CLL prognosis was validated in an additional patient cohort.12 Interestingly, thyroid hormone receptor-associated protein 3 (TRAP3), T-cell leukemia/lymphoma protein 1A (TCL1A), protein S100A8, and myosin-9 have been reported to significantly predict the prognosis of CLL.12
Given the complex nature of proteomics, in our previous study a larger effort was made for the proteomics-based discovery of CLL-related proteins as opposed to the validation of the impact of those proteins on CLL prognosis.12 Transcriptomics data sets that are available from public repositories, such as Gene Expression Omnibus (GEO)14 and The Cancer Genomics Atlas (TCGA),15 represent rich resources of information that can be used to investigate the relevance of a transcript expression to a disease. Therefore, the goal of this study was to use independent transcriptomics data sets of cancer patients with prognostic information from public repositories to validate the relevance of our previously described CLL-related proteins at the level of transcription (mRNA) to the prognosis of CLL.
Methods
Study design
The present work is a validation study that was based on the use of transcriptomics data sets of cancer patients, which are publicly available from GEO and TCGA, in order to confirm the relatedness of our previously described CLL-related proteins at the level of mRNA to the prognosis of CLL. This study was ethically approved by the Ethical Committee of the Deanship of Scientific Research, Majmaah University (Approval No: MUREC-July.02/COM-2018/8) and was conducted at Majmaah University, Al Majmaah, Kingdom of Saudi Arabia between January 2017 and July 2018.
Inclusion and exclusion criteria
A number of criteria were applied for the search of transcriptomics data sets of CLL from GEO that would be used for the validation analyses. All CLL transcriptomics data sets that did not contain clinical details about the prognosis of individual patients or were based on insufficient number of patients, which prevented reaching a firm statistical conclusion of the validation analyses, were excluded. In contrast, for transcriptomics data sets of CLL to be included in this study they had to pass 3 inclusion criteria. First, data sets must have contained clinical details about CLL prognosis, such as time-to-first treatment (TTFT) or overall survival (OS), for the individual patients whose samples were studied. Second, data sets had to be generated from sufficient number of patients to enable drowning a definitive conclusion of the validation analyses (number of patients per data set ≥100). Different data set had to be reported by independent research groups using the same platform of oligonucleotide microarray.
Transcriptomics data sets from GEO
Two transcriptomics data sets of CLL were found based on the inclusion and exclusion criteria (GEO accession number: GSE39671 and GSE22762).16,17 The data set GSE39671 contained information of TTFT and the data set GSE22762 included details of OS for the individual patients. Both data sets were reported by independent authors and were based on Affymetrix Human Genome U133 Plus 2.0 Array (USA). The data set GSE39671 was generated from 130 CLL patients and the data set GSE22762 was reported from 107 CLL patients.
The DataSet SOFT files of the transcriptomics data sets were downloaded from GEO. Then, g:Profiler and retrieve/ID mapping tool with the UniProt database were used to cross-reference the ID references (probe IDs) of Affymetrix Human Genome U133 Plus 2.0 Array with the corresponding UniProt entry identifiers (protein-specific identifier).18-20 Next, the UniProt entry identifiers of our CLL-related proteins were used to identify the corresponding transcripts in the 2 transcriptomics data sets.
Transcriptomics data sets from TCGA
Independent transcriptomics data sets of various types of cancer with available prognostic data, such as OS or relapse free survival (RFS), that were generated and published by the TCGA research network were used.15 These data sets were employed to further investigate the relevance of HNRNPUL2 to the prognosis of malignancies other than CLL. The analyses were conducted using cBioPortal and Onco Query Language (OQL), the combination of which allows users to determine if a particular value of gene expression can segregate patients into 2 groups with different prognoses.21 Heterogeneous nuclear ribonucleoprotein U like 2 “HNRNPUL2: EXP>x” was the OQL that was applied to the transcriptomics data sets to separate patients in each data set into 2 groups (a low-expression group with HNRNPUL2 expression below “x” and a high-expression group with HNRNPUL2 expression above “x”), “x” is a value of z score that varied in each data set. Details of the transcriptomics data sets (n=6 independent data sets) and the applied OQL, through which HNRNPUL2 exhibited prognostic importance in the present study, are summarized in Table 1.
The TCGA transcriptomics data sets and OQL through which HNRNPUL2 exhibited a prognostic significance.
Pathway-enrichment analyses
To gain insights into the pathways to which the transcripts of interest are assigned, pathway-enrichment analyses were conducted using a curated pathway database “Reactome”.22 The analyses were restricted to human specific pathways using the tool “Analyze Data”. Reactome reports enriched pathways by a factor of p-value, which indicates the probability of a pathway being identified by chance. In addition, Reactome reports the false discovery rate (FDR) of a corrected enrichment probability. Together, the p-value and the FDR provide accurate measures of false identification of a pathway.22 In the present study, only pathways that were significantly enriched (p≤0.05 and FDR ≤0.05) were reported.
Statistical analyses
Prism Graphpad software was used to create Kaplan-Meier curves of TTFT, RFS, and OS; the Log-rank test was used to calculate p-values and hazard ratios (HRs). Excel software was employed for the correlation analyses and calculation of Pearson scores (PS). The p-values and the FDRs of the pathway-enrichment analyses were calculated using the Reactome pathway knowledge base.22 A heatmap visualization of the correlation analyses was constructed using the heatmapper web-based tool.23
Results
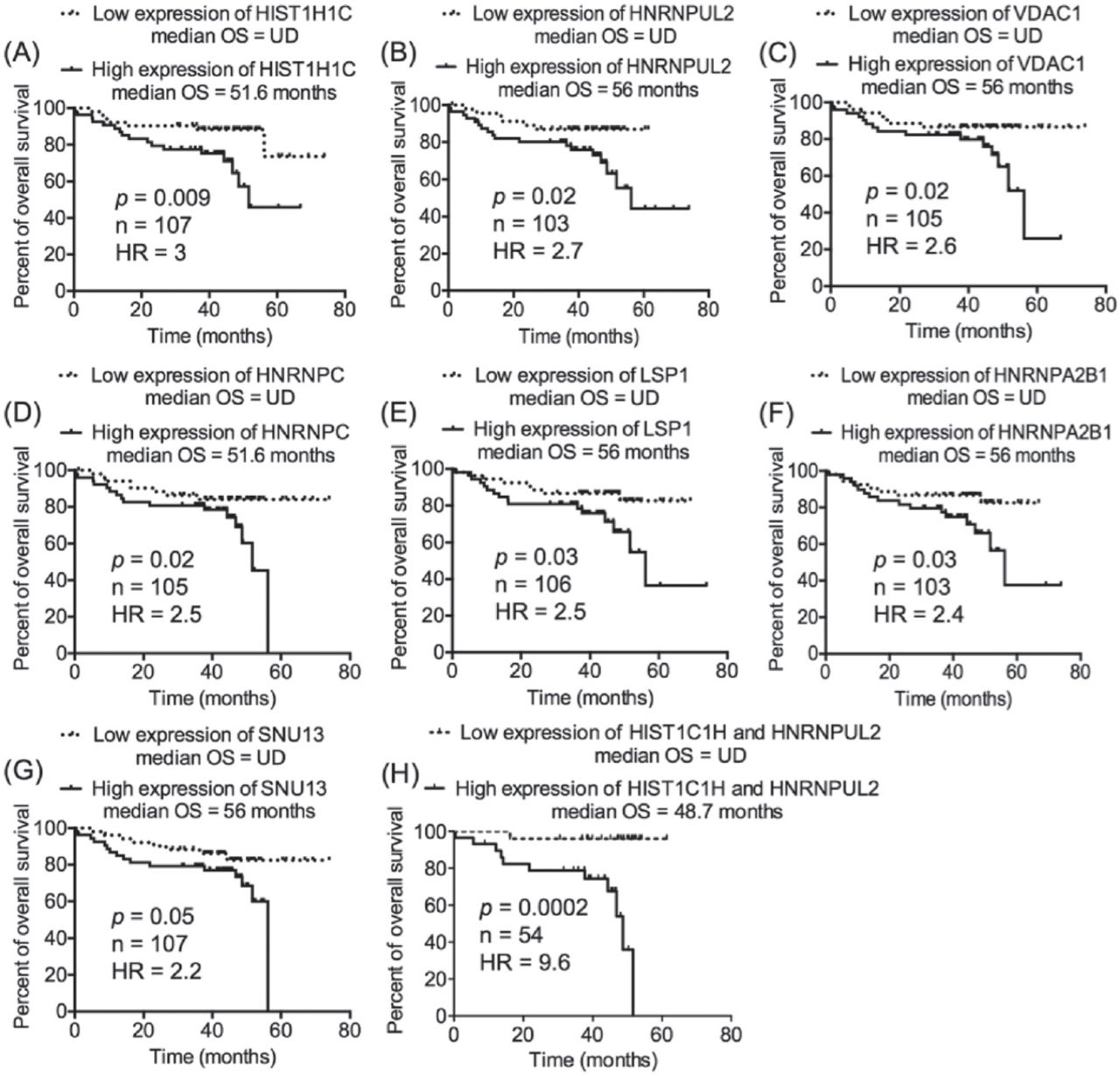
Our previous work on CLL proteomics described 63 candidates as CLL-related proteins, of which TRAP3, TCLA1, S100A8, and myosin-9 were further studied and were found to significantly predict the prognosis of CLL.12 In the present study, the transcript expression of the remaining CLL-related proteins, whose prognostic value was not validated in our previous study (n=59), were investigated in the context of CLL prognosis. The transcriptomics data set GSE39671 contains data regarding TTFT (n=130), and the transcriptomics data set GSE22762 included information of OS (n=107).16,17 Therefore, the 2 transcriptomics data sets were used independently to validate the relevance of the 59 CLL-related proteins at the level of transcription (mRNA) to CLL prognosis (TTFT and OS). The patients were divided into 2 groups (a low-expression group and a high-expression group) based on the median expression of the corresponding transcripts to the proteins of interest. This step was conducted separately on each one of the 2 transcriptomics data sets and for each one of the transcripts of interest. Next, TTFT and OS of the low-expression and high-expression groups were compared using Kaplan-Meier curves. Interestingly, the validation analyses revealed that the cognate transcripts of 9 proteins of TTFT and 7 proteins of OS were significantly predictive in CLL patients (Figures 1 & 2). Of these transcripts, 2 (HNRNPUL2 and HIST1H1C) significantly predicted an early need for therapy in the transcriptomics data set GSE3967116 and short OS in the transcriptomics data set GSE2276217, increasing the validity of their prognostic significance in CLL. Furthermore, of the 14 transcripts, 8 corresponded to different types of heterogeneous nuclear ribonucleoproteins (HNRNPs), indicating a role of such molecules in the prognosis of CLL.
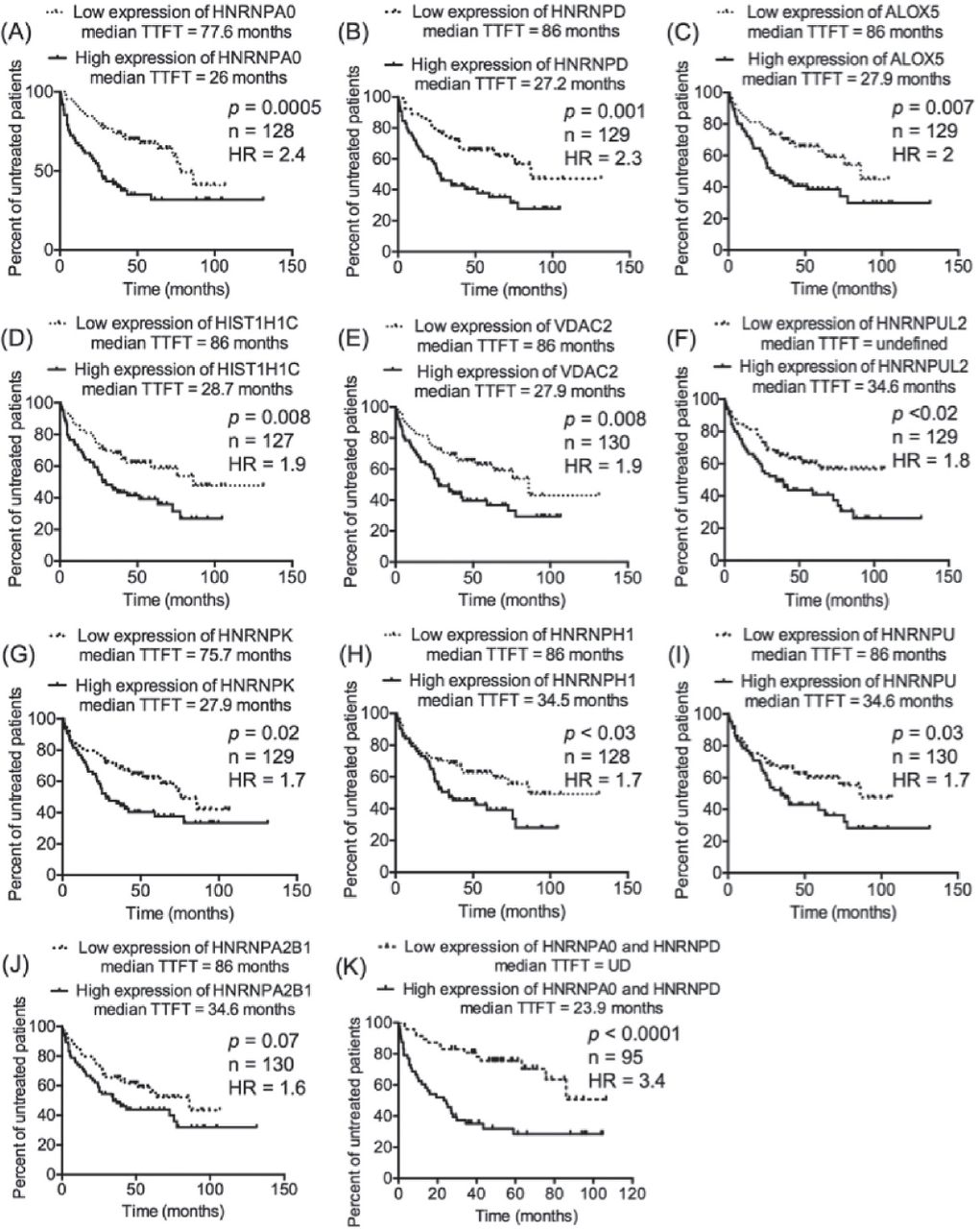
Nine of the chronic lymphocytic leukemia (CLL) -related proteins had transcript expression that predicted time-to-first treatment (TTFT) in CLL patients. The median expression of the transcripts of interest was used to divide CLL patients into 2 groups: low-expression group, in which the expression of a transcript of interest was smaller than its median, and high-expression group, where the expression of a transcript of interest was greater than its median. This step was carried out independently for each transcript of interest. A-J) The TTFT in the low expression and high expression groups was compared using Kaplan-Meier curve. K) Patients with discordant expression of HNRNPA0 and HNRNPD were not included in the analysis.
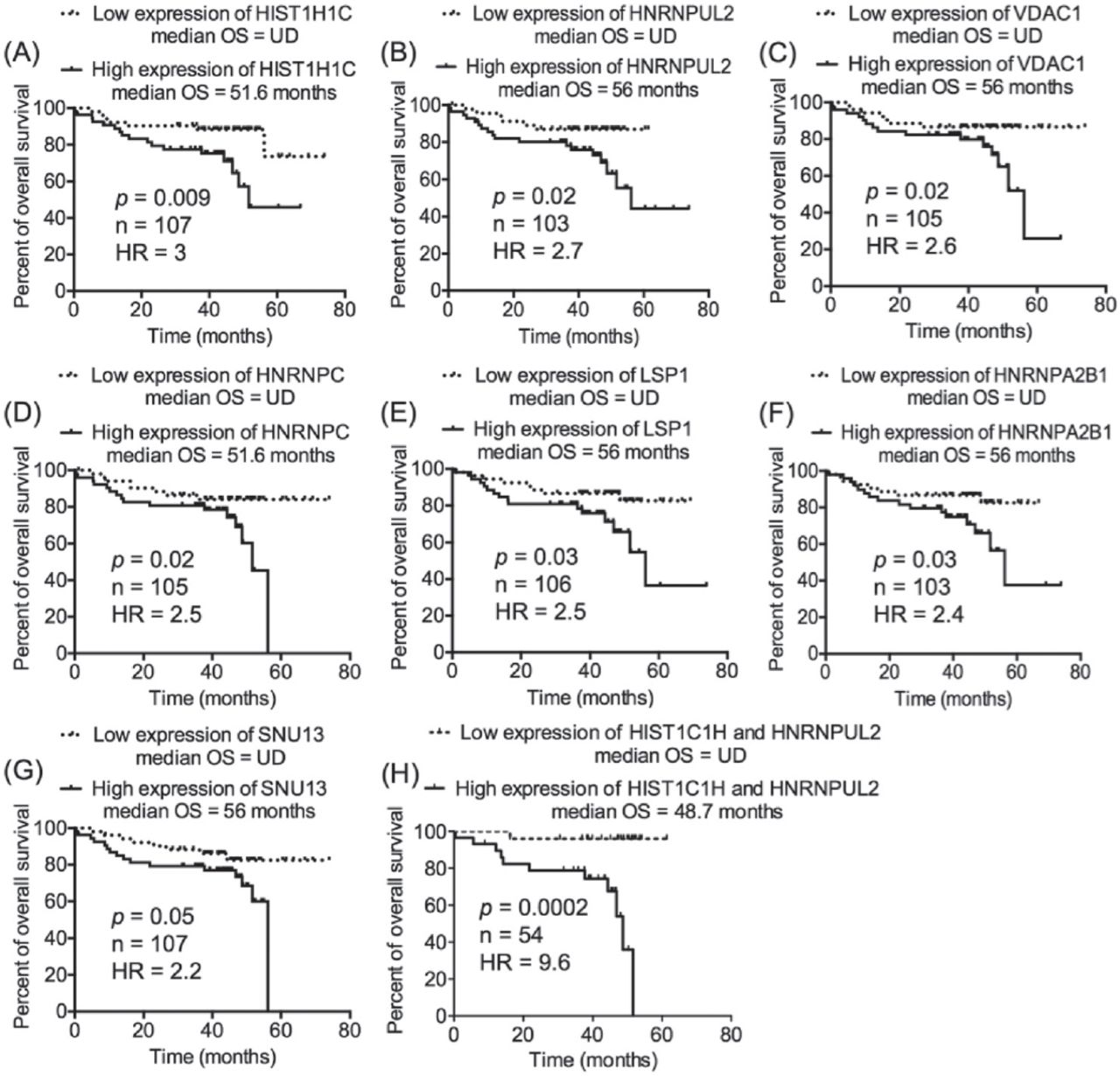
Seven of the chronic lymphocytic leukemia (CLL)-related proteins processed transcript expression that predicted overall survival (OS) in CLL patients. Chronic lymphocytic leukemia patients were divided into 2 groups based on the median expression of the transcripts of interest; low-expression group (transcript expression <median expression) and high-expression group (transcript expression >median expression). This step was performed independently for each one of the transcripts of interest. A-G) Kaplan-Meier curve was utilized to compare the OS of the low-expression and high-expression groups. H) Patients with discordant expression of HIST1C1H and HNRNPUL2 were not included in the analysis.
Among the 9 transcripts that predicted TTFT in the transcriptomics data set GSE3967116, HNRNPA0 and HNRNPD were the best indicators of early therapy (HR=2.4 [Figure 1A] and HR=2.3 [Figure 1B]). Combining HNRNPA0 with HNRNPD improved the prediction of TTFT and increased the HR to 3.4 (Figure 1K). Likewise, combining HNRNPUL2 with HIST1C1H dramatically improved the prediction of OS in CLL patients of the transcriptomics data set GSE2276217; the HR was 9.6 of the combined HNRNPUL2 with HIST1C1H (Figure 2H) compared with 3.0 for HIST1C1H (Figure 2A) and 2.7 for HNRNPUL2 (Figure 2B).
Next, pathway-enrichment analyses using Reactome database were conducted for the 14 transcripts that predicted the prognosis of CLL. Three pathways were reported: mRNA splicing (p=5.23×10-9, FDR=1.42×10-7), processing of capped Intron-containing pre-mRNA (p=2.74×10-08, FDR=4.65×10-07), and gene expression (p=0.0004, FDR=0.004). Interestingly, the mRNA splicing pathway was enriched by the 8 different types of HNRNPs.
Of the 8 HNRNPs that predicted the clinical outcomes of CLL, increased expression of HNRNPUL2 significantly identified patients with poor prognosis of CLL in the 2 independent transcriptomics data sets (GSE39671 and GSE22762).16,17 In an attempt to explain this finding, correlation analyses using Pearson score were conducted on the CLL transcriptomics data set (GSE39671; n=130) in order to identify genes whose expression correlated with the expression of HNRNPUL2. From the transcriptome of CLL cells, 1171 genes exhibited an expression that significantly correlated with the expression of HNRNPUL2 (Pearson score ≥0.50; p<0.00001) in 130 patients. To gain insights into the function of these genes, they were subjected to pathway-enrichment analyses using Reactome database. Table 2 lists the CLL-related pathways that were significantly enriched by the 1171 genes. Figure 3A shows a heatmap presentation of the correlation between the expression of the genes that enriched cell cycle pathway and the expression of HNRNPUL2 in 130 patients. The correlation analyses also reported known important genes in the pathology and prognosis of CLL, such as apoptosis regulator (BCL-2), apoptosis inhibitor 5 (API5), and oncogene DEK, that significantly correlate with the expression of HNRNPUL2 (Figure 3B).
Pathway-enrichment analyses of the genes that correlated with HNRNPUL2 in CLL patients.

Heatmap presentation of the correlation between HNRNPUL2 and genes of interest in chronic lymphocytic leukemia (CLL) patients. The interrogation of Reactome database reported a significant enrichment of the cell cycle pathway by 101 genes of the genes whose expression significantly correlated with the expression of HNRNPUL2 in the CLL transcriptomics data sets (GSE39671; n=130). Excel software was used to sort the 130 CLL patients from left to right based on the ascending expression HNRNPUL2 (from lowest expression to highest expression). Then, the 101 genes were sorted from top to bottom on the bases of their Pearson scores (from 0.74-0.50), with HNRNPUL2 being at the top of the list. A) Heatmapper was used to construct a heatmap graphic based on the expression of the 101 cell cycle gens and HNRNPUL2 in the 130 patients. B) Of the other genes that correlated with the expression of HNRNPUL2, 7 (Pearson score ranged from 0.74-0.50) were previously shown to drive the progression of CLL.
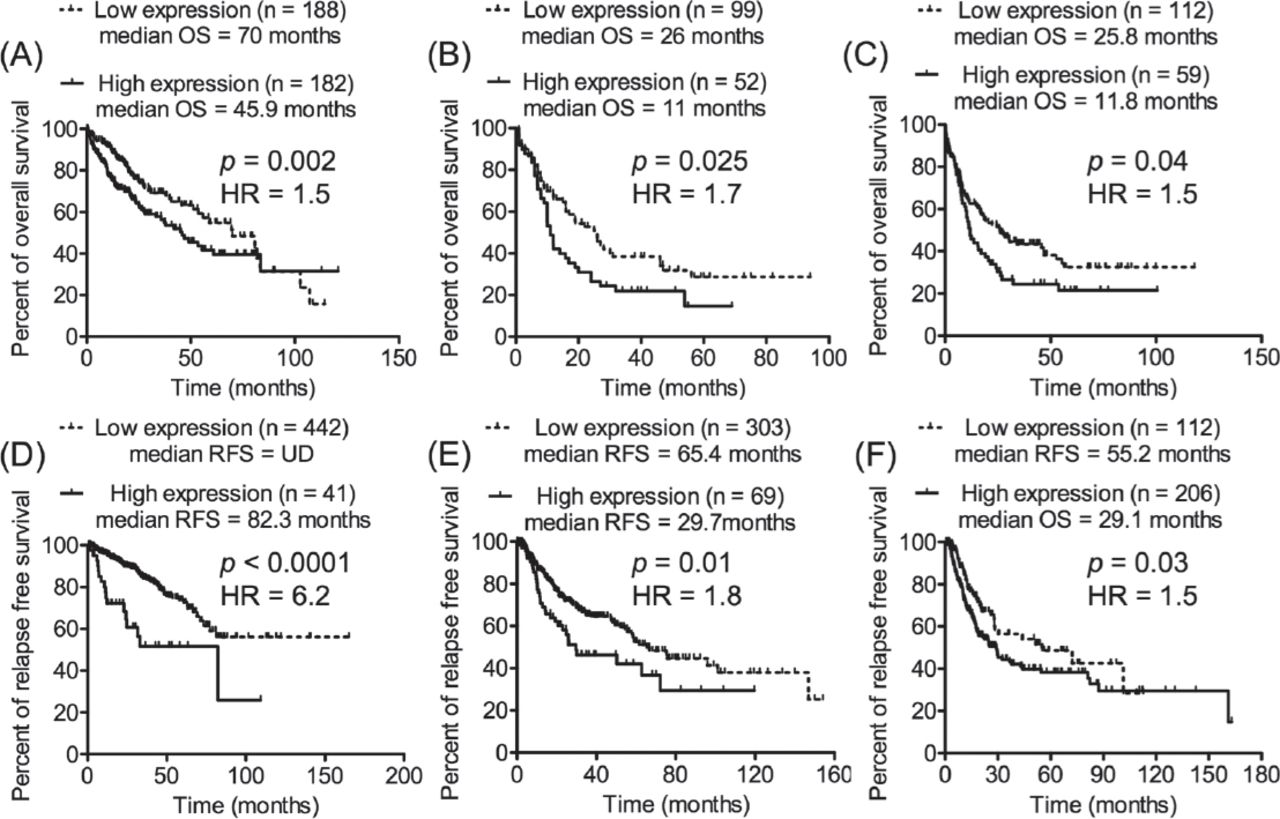
Next, investigations were performed to determine whether the expression of HNRNPUL2 possessed prognostic importance in malignant diseases other than CLL. The Cancer Genomics Atlas (TCGA) transcriptomics data sets of different types of cancer with clinical information about OS or RFS and the cBioPortal with OQL were utilized. Initially, the median expression of HNRNPUL2 in the TCGA transcriptomics data sets was used to divide cancer patients in each data set into 2 groups (low-expression and high-expression groups). Next, the Kaplan-Meier curve was used to compare the OS or RFS data of the 2 groups of patients. The analyses revealed that the median expression of HNRNPUL2 failed to exhibit prognostic significance. Therefore, an effort was made using the OQL to determine if an expression value of HNRNPUL2 (reported as a value of standard deviation from a mean: z score) that separates cancer patients into 2 groups with different prognoses could be found in the used TCGA transcriptomics data sets. Consequently, an increased expression of HNRNPUL2 based on different z scores was found to significantly identify a subset of cancer patients with short OS or early relapse in 6 independent transcriptomics data sets of various types of cancer (Figure 4).
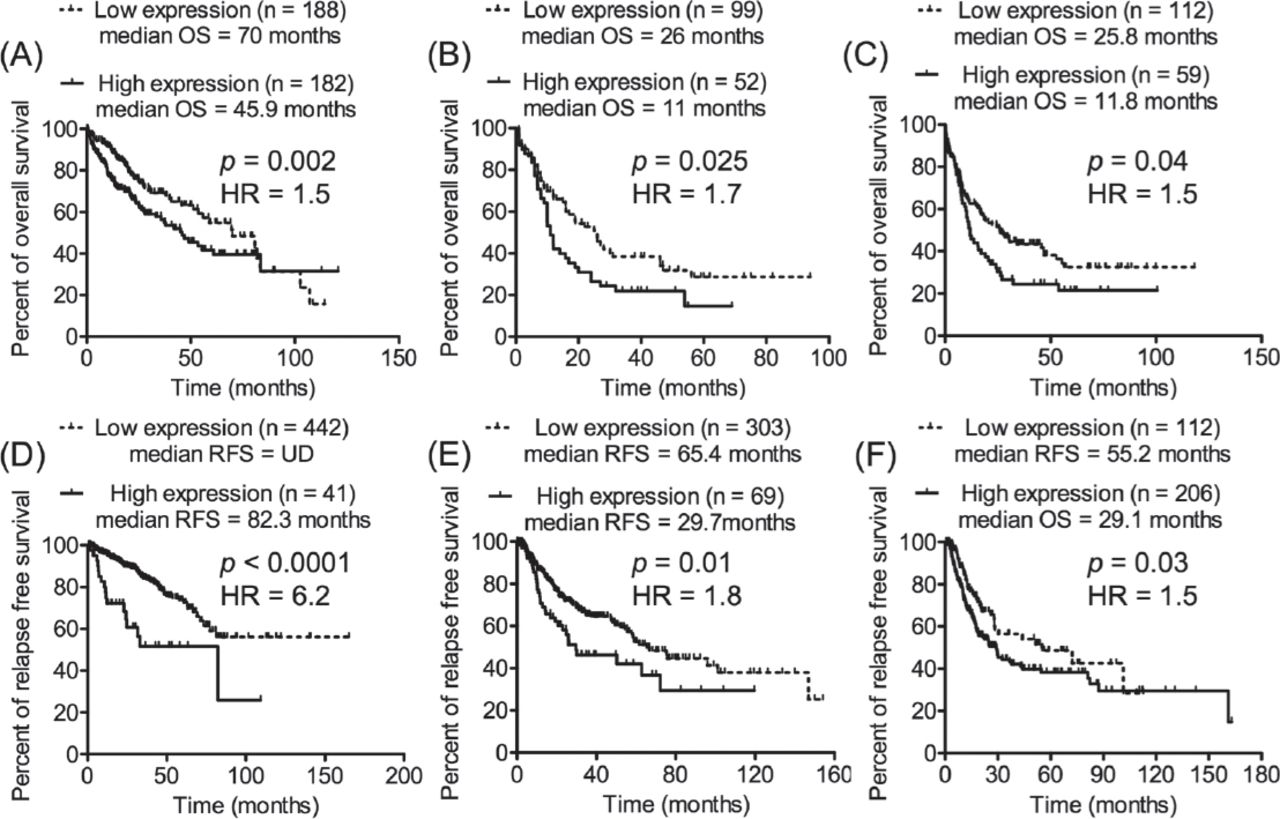
Increased expression of HNRNPUL2 was indicative of poor prognosis of different types of cancer. The prognostic potential of HNRNPUL2 was assessed in independent transcriptomics data sets of various malignancies from TCGA using cBioPortal and OQL. Different z scores of HNRNPUL2 expression were applied on the TCGA transcriptomics data sets to divide patients into 2 groups; low expression group (HNRNPUL2 expression <z score) and high expression group (HNRNPUL2 expression >z score). Increased expression of HNRNPUL2 was found to significantly predict short overall survival (OS) or short relapse free survival (RFS) in 6 independent transcriptomics data sets of different types of cancer. A) Liver hepatocellular carcinoma (TCGA, Provisional) with z score = -0.12. B) Acute myeloid leukemia (TCGA, Provisional) with z score = 0.413. C) Acute myeloid leukemia (TCGA, NEJM 2013) with z score = 0.38. D) Prostate adenocarcinoma (TCGA, Provisional) with z score = 1.4. E) Lung squamous cell carcinoma (TCGA, Provisional) with z score = 1. F) Bladder urothelial carcinoma (TCGA, Provisional) with z score = -0.4. OQL - onco query language, TCGA - The Cancer Genomic Atlas.
Discussion
In the present study, the cognate transcripts of 14 of our CLL-related proteins,12 were found to significantly predict the clinical outcomes of CLL. These transcripts may be accordingly considered good candidate to serve as prognostic markers of CLL. Interestingly, 8 of the 14 transcripts were different types of HNRNPs, and HNRNPUL2 was also reported to predict the prognosis of various types of cancer in addition to CLL. Although HNRNPs have been implicated in a wide range of neoplasms, they have not been linked to the prognosis of CLL. Overexpression of HNRNPA2/B1 was documented in malignant tissues of different organs including breasts, livers, lungs, and pancreas.24 Furthermore, HNRNPK is overexpressed in lung cancer and liver cancer and predicts poor prognoses of head and neck carcinoma, oral squamous cell carcinoma, acute myeloid leukemia, and T-cell leukemia/lymphoma.25 Similarly, HNRNPD is associated with esophageal squamous cell carcinoma and indicates an aggressive type of the disease.26 Collectively, the prognostic significance of HNRNPs in CLL shown in the current work supports the previously reported role of HNRNPs in cancer prognoses.
The interrogation of the Reactome database revealed that of the 14 transcripts whose increased expression predicted a poor prognosis of CLL, 8 different types of HNRNPs significantly enriched the mRNA splicing pathway. In agreement with this finding, HNRNPs have been commonly implicated in alternative splicing that favors the survival of malignant cells. For example, in acute T-cell leukemia cells, HNRNPA2/B1 promotes the production of the anti-apoptotic isoform of DnaJ protein Tid1 (Tid1-S) over the synthesis of the pro-apoptotic isoform (Tid1-L), supporting the survival of leukemic cells.27 In addition, HNRNPK has been shown to negatively regulate the transcription of the pro-apoptotic splice isoform of BCL-X (BCL-Xs) in prostate cancer cells and cervical cancer cells.28 In cervical cancer cells, HNRNPC positively regulates the exclusion of the FAS exon 6 and promotes the expression of the anti-apoptotic splice isoform.29 In CLL, altered splicing as evidenced by an increased expression of spliceosome components including HNRNPs was implicated in the tumorgenesis of the disease.30 The positive impact of HNRNPs on the survival of cancer cells exerted through their roles in alternative splicing suggests an explanation of the significant prediction of the aggressive form of CLL by the increased expression of HNRNPs. Furthermore, these findings provide a rationale for targeting HNRNPs to antagonize the survival of CLL cells.
Of the 8 HNRNPs that predicted the prognosis of CLL, increased expression of HNRNPUL2 identified a subset of patients with short survival and early need for therapy in the 2 independent transcriptomics data sets of CLL. The aggressive form of CLL is characterized by active pathways that promote cellular proliferation and survival, such as cell cycling, NF-κB,32 BCR signaling, and response to hypoxia.31,33,34 Interestingly, these pathways were significantly enriched by the genes that exhibited a significant correlation with the expression of HNRNPUL2 in 130 patients. Furthermore, genes that are known to support the survival of CLL cells such as API5,35 BCL2,36 and oncogene DEK,37 were also found to significantly correlate with the expression of HNRNPUL2. These findings suggest that increased expression of HNRNPUL2 marks CLL cells with active proliferation and augmented survival, which fits with the currently described role of HNRNPUL2 as a poor prognostic marker of CLL. These data also point out to the possibility of HNRNPUL2 to serve as therapeutic target in CLL cells.
Heterogeneous nuclear ribonucleoproteins belong to a big family of related proteins that are highly abundant in human cells.38 Therefore, HNRNPs are less challenging to identify using proteomics approach; in our previous study we reported 12 HNRNPs as CLL-related proteins.12 As mentioned earlier, HNRNPs have been implicated in various kinds of cancer including CLL. These factors perhaps have favored HNRNPs in contrast with the other CLL related proteins to be prognostically important.
A number of points should be considered while viewing the present findings. First, this study shows the usefulness of transcriptomics data set from GEO and TCGA for investigating the relevance of a protein to a disease by examining the expression of the protein’s corresponding transcript in relation to a disease prognosis.39 However, the findings obtained following such a method should be interpreted with caution because protein expression does not always correlate with transcript expression.40 For example, although increased expression of the HNRNPs significantly predicted a poor prognosis of CLL in the current study, these findings do not necessarily indicate a significant association of the HNRNPs (as proteins) with the aggressive form of the disease. As a result, the prognostic value of the HNRNPs (as proteins) in CLL remains to be investigated. Second, transcriptomics findings of interest are traditionally validated using real-time polymerase chain reaction (RT-PCR); therefore, measuring the expression of the 14 transcripts in CLL samples using RT-PCR is worthwhile to confirm the expression patterns of these transcripts. Third, cohort-to-cohort variations in terms of disease characteristics and therapy are likely to happen. Therefore, examining the prognostic potential of the 14 transcripts in additional CLL cohorts is required to further validate the utility of these biomarkers across CLL patients with different diseases characteristics and types of treatment. Forth, the clinical usefulness of the current prognostic markers compared with the common prognostic markers of CLL was not explored due to the unavailability of the latter in the 2 transcriptomics data sets of CLL. Therefore, it would be interesting to determine whether the 14 transcripts provide additional prognostic information to what can be obtained by the commonly applied prognostic markers of CLL.
In conclusion, 2 independent transcriptomics data sets of CLL from GEO were used to validate the relevance of our CLL-related proteins at the level of mRNA to CLL prognosis. The cognate transcripts of 14 of these proteins significantly predicted the clinical course of CLL; hence, they may have the potential to serve as prognostic markers of the disease. In 14 transcripts, HNRNPUL2 was also found to be informative of poor prognosis of different neoplasms other than CLL in 6 independent transcriptomics data sets from TCGA. Interestingly, the correlation analyses and the interrogation of the Reactome database have yielded an explanation for the prognostic value of HNRNPUL2 and gave a rationale for targeted therapy of CLL through targeting HNRNPUL2. Additional investigations of the 14 transcripts in parallel with the common prognostic markers of CLL using a cohort of CLL patients is required to further assess the clinical usefulness of the 14 transcripts as prognostic markers. The present study also calls for further investigations on HNRNPs in the context of targeted therapy of CLL.
Acknowledgment
The authors gratefully acknowledge the American Manuscript Editors (www.americanmanuscripteditors.com) for English language editing.
Footnotes
Disclosure. The author has no conflict of interests, and the work was not supported or funded by any drug company.
- Received January 20, 2019.
- Accepted February 27, 2019.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.