


Abstract
Chronic lymphocytic leukemia (CLL) is an incurable malignant disease of B-lymphocytes characterized by drastically heterogeneous clinical courses. Proteomics is an advanced approach that allows a global profiling of protein expression, providing a valuable chance for the discovery of disease-related proteins. In the last 2 decades, several proteomics studies were conducted on CLL to identify aberrant protein expression underpinning the malignant transformation and progression of the disease. Overall, these studies provided insights into the pathology and prognosis of CLL and reveal protein candidates with the potential to serve as biomarkers and/or therapeutic targets of the tumor. The major findings reported in these studies are discussed here.
Chronic lymphocytic leukemia (CLL) is an adult hematologic cancer characterized by a significant expansion of small, mature-looking B-cells, in peripheral blood and lymphoid tissues.1 Although the key-cause of CLL yet to be identified, some genetic variants have been found to elevate the risk for developing the disease.2 The prognosis of CLL is extremely variable; whereas some patients required an early therapy and show poor-treatment response leading to short survival (poor prognosis), other patients show no or late need of therapy and have long overall survival (good prognosis).3 In our previously published work, we reviewed the key molecular drivers of CLL that have major roles in determining the clinical outcomes of the cancer.4 Some of these drivers have an established prognostic importance in CLL; hence, they have been commonly used as biomarkers of the disease prognosis.4 For example, the mutational status of immunoglobulin heavy variable genes (IGHV), which code for components of B-cell receptor (BCR), is informative of CLL prognosis; un-mutated IGHV (UM-CLL) is associated with an unfavorable prognosis, whereas mutated IGHV (M-CLL) denotes a desirable prognosis.5 In addition, a high expression of CD38, 70 kDa zeta-associated protein (ZAP-70), C-X-C chemokine receptor type 4 (CXCR4), and CD49d is predictive of poor prognosis.6-8 Chromosomal aberrations, such as deletions in 11q and 17p are also characteristics of adverse prognosis.9 However, deletions in 13q are indicative of good prognosis.9 Recent modalities of CLL therapy lessen the burden of the tumor and increase the overall survival.10 Nevertheless, CLL remains incurable and is life-threatening for many patients, especially those with poor prognosis.10
Proteomics is an advanced approach that enables large-scale characterizations of different aspects of proteins, such as protein expression profiling, post translation modification (PTM), protein localization, and protein function.11 Standard proteomics studies rely on 4 major steps; reduction of proteins/peptides complexity by separation means like gel electrophoresis and liquid chromatography, determination of peptides or peptide fragments masses using mass spectrometry, use of a protein database and bioinformatics tool for the interpretation of the mass spectrometry data into protein identification.12 Although genetic predisposition provides explanations, at least partially, for the heterogeneous prognosis of CLL, aberrant protein production has been reported to significantly influence the disease behavior and clinical outcomes.2 For example, the migratory proteins C-X-C chemokine receptor type 4 (CXCR4) and CD49d; the anti-apoptotic proteins induced myeloid leukemia cell differentiation protein Mcl-1 and apoptosis regulator Bcl-2; have been shown to heavily contribute to the worse prognosis of the disease.4 These proteins were reported using single protein detection approaches. Proteomics, on the other hand, allows the identification and quantification of a large number of proteins simultaneously.11 Therefore, proteomics is a very useful approach for the global characterization of differentially expressed proteins in CLL to provide new insights into the disease and reveal potential biomarkers for diagnosis/prognosis and targets for therapy.13 In the present review, the findings reported by the studies that conducted proteomics-based protein expression profiling on clinical CLL samples will be discussed.
Differential proteome is associated with UM-CLL
As mentioned earlier, BCR is considered one of the main drivers of CLL progression (Figure 1). As a result, characterizing the proteomics signature of UM-CLL and M-CLL has the potential to improve our understanding of the molecular variabilities between the 2 forms of the disease and highlight potential prognostic markers and therapeutic targets. Following the same concept, Cochran et al used 2-dimensional electrophoresis (2DE) and mass spectrometry to perform proteomics investigations on CLL samples from 12 patients; 6 UM-CLL and 6 M-CLL.14 The authors reported 3 proteins with differential expression; F-actin-capping protein beta subunit and laminin-binding protein precursor showed reduced expression in UM-CLL samples relative to M-CLL samples. These proteins play important roles in the organization and activation of the cytoskeleton, suggesting an altered cytoskeleton activity in the aggressive form of CLL (UM-CLL). Another interesting protein is nucleophosmin, which was undetectable in the UM-CLL cases, but was identified in all the M-CLL samples. This finding was further supported by a later study that reported an active metabolism and degradation of nucleophosmin in UM-CLL.15 Nucleophosmin augments the stability and function of the tumor suppressor protein p53.16 Defects of p53 have been associated with a rapid progression of CLL and undesired prognosis.17 Therefore, the specific expression of nucleophosmin in M-CLL may contribute to a better prognosis of the disease through its positive impact on p53. Given the specific expression of nucleophosmin in M-CLL, it merits further validation in a larger cohort of patients to confirm its utility as a good prognostic marker of CLL.
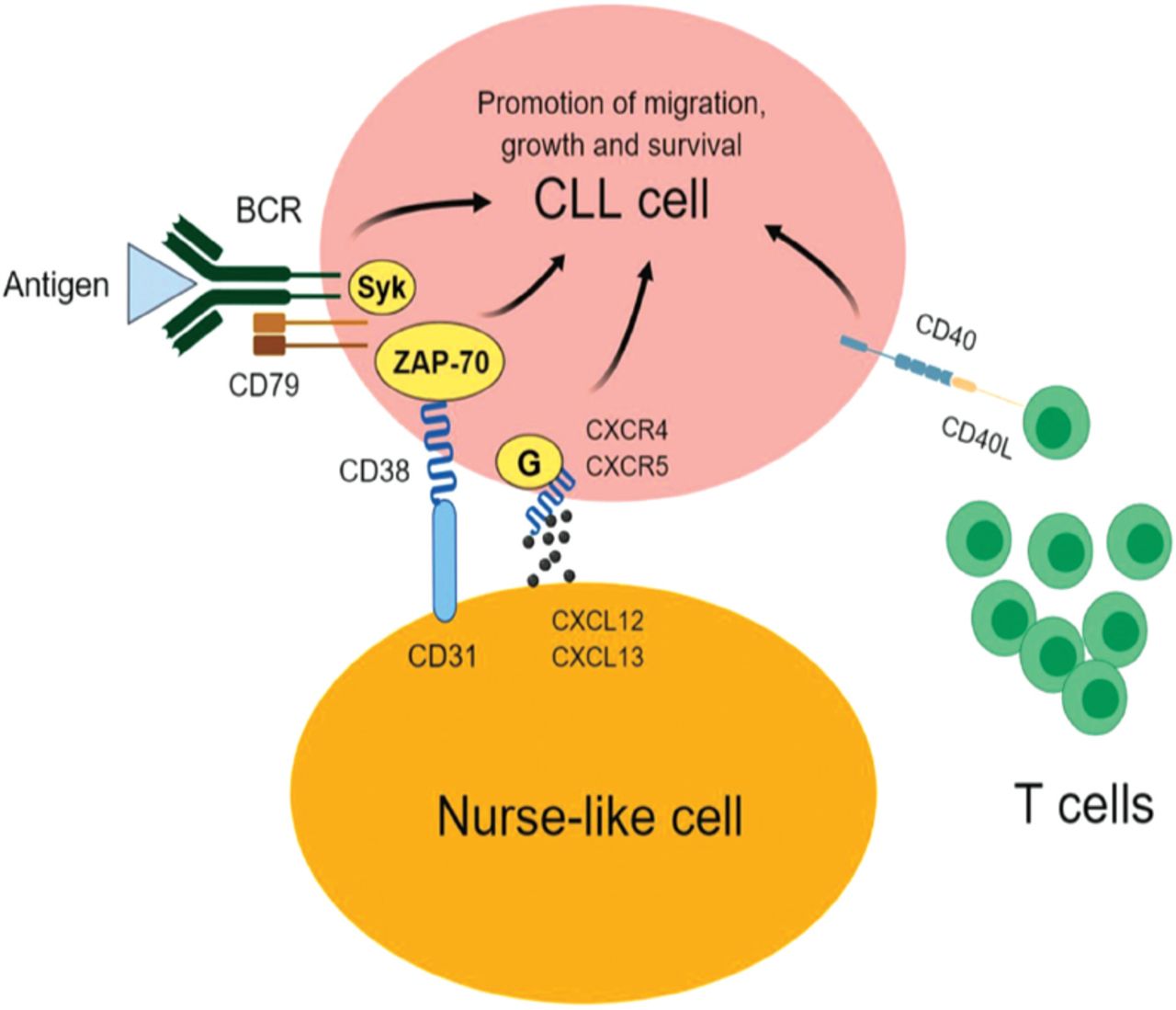
The main drivers of chronic lymphocytic leukemia (CLL). The engagement of B-cell receptor (BCR) on CLL cells with an antigen provides pro-survival and proliferation signals that are transduced to the nucleus by various proteins including spleen tyrosine kinase (SYK) and/or 70-kDa zeta-associated protein (ZAP-70). C-X-C chemokine receptor type 4 (CXCR4) on CLL cells binds with its ligand C-X-C motif chemokine 12 (CXCL12; also known as stromal cell-derived factor 1) causing the malignant cells to migrate to bone marrow and close proximity to nurse-like cells (NLC). CD31 on the surface of NLC also interacts with the CD38 on CLL cells, resulting in the proliferation of CLL cells. The engagement of CD40 on CLL cells with CD40L on T-cells promotes the growth and survival of the cancerous cells.
Employing cleavable isotope-coded affinity tags (cICAT) and one-dimensional liquid chromatography (1DLC) coupled with tandem mass spectrometry, Barnidge et al searched for differentially expressed proteins in CLL cells from 2 patients (UM-CLL and M-CLL).18 The investigations were conducted on 2 different protein extracts; cytosolic proteins extract and membrane proteins extract. A CLL proteome consisting of 326 proteins in the cytosolic extracts and 212 proteins in the membrane extracts was reported, of which 13 proteins exhibited differential expression in the 2 samples. Following validation analyses of the altered expression in 12 CLL samples, cytochrome C oxidase subunit 6B1 (COXG, also known as COX6B1) was reported with low-expression in UM-CLL relative to M-CLL. Cytochrome C oxidase subunit 6B1, which was specifically detected in the membrane extracts, is a mitochondrial enzyme involved in the mitochondrial respiratory chain, thus this finding provides evidence of an altered mitochondrial protein expression as a discriminator between UM-CLL and M-CLL. The relationship of COXG with cancer was also reported at the genomics level; 2 single nucleotide polymorphisms (SNPs) of COXG were found to be predictive of poor prognosis of colorectal cancer.19 Taken together, these findings support the implication of altered mitochondrial function in cancer pathogenesis.20
In a more comprehensive study, Eagle et al21 used isobaric tags (iTRAQ) with 2-dimensional liquid chromatography (2DLC) connected to tandem mass spectrometry in order to describe a proteome signature associated with UM-CLL as opposed to M-CLL. The study was conducted on 9 UM-CLL samples and 9 M-CLL samples; and the researchers reported a CLL proteome containing 3521 proteins, from which 274 had altered abundance in the 2 subsets of samples. Pathway enrichment analysis of these proteins identified migration defects in UM-CLL cells. For instance, integrin chains (alpha-L and beta-2), which are important proteins for the migration of CLL cells into lymph nodes,22 RAS guanyl-releasing protein 2, which plays roles in the activation of integrin,23 and talin, which is required for the proper function of integrin,22 were significantly down-regulated in UM-CLL cells. Migration of CLL cells to lymph nodes is essential for the growth and survival of the tumor cells; therefore, UM-CLL was shown to compensate for the impaired integrin (alpha-L and beta-2)-mediated migration by elevating the expression of the other integrin chains (alpha-4 and beta-1).24-26 The proteomics findings also showed that UM-CLL cells were characterized by increased adhesion as evidenced by an elevated expression of CD44, which is necessary for the adhesion of CLL cells within the lymph node microenvironment;27 and decreased egress of the malignant cells from lymph nodes as demonstrated by a reduced expression of adenylate cyclase-inhibiting G alpha protein and dynamin-2. High-adhesion and limited passage from lymph node to peripheral blood are 2 factors known to feature an extended retention of CLL cells in the tumor-supportive microenvironment of lymph nodes, which explain, at least partially, the aggressive nature of UM-CLL.26 In line with this view, Eagle et al also reported a significant association between UM-CLL and lymphadenopathy despite a similar disease burden in both subsets of the studied samples (UM-CLL and M-CLL).21 In addition to the adhesion property of CD44, it also protects CLL cells from apoptosis and promotes the disease progression,28 providing further explanation for the poor prognosis associated with UM-CLL. In agreement with the findings reported by Cochran et al,14 UM-CLL cells were also shown by Eagle et al21 to decrease the expression of proteins that are involved in cytoskeleton activity, such as actin cardiac muscle 1a, actin cytoplasmic 2, actin-related protein 3 and filamin-A. These data stress on the aberrant cytoskeleton activity as a characteristic of UM-CLL.
As mentioned earlier, determining the mutational status of IGHV is essential to predict the clinical outcomes of CLL.4 This step is routinely performed using DNA sequencing technologies.3 On the other hand, Díez et al proposed a novel strategy using tandem mass spectrometry to conduct peptide sequencing of surface immunoglobulin (sIg) and other immune-related proteins of CLL cells.29 This approach is complementary to DNA sequencing of IGHV using molecular biology methods. However, the advantage of Díez’s method is that it allows peptide sequencing of sIg as well as other CLL-related proteins in a single assay, providing more information about the proteins that determine the behavior of CLL.29
Proteomics insights into BCR signaling of CLL
B-cell receptor signaling is a key event for the progression of CLL; stimulation of BCR was reported to promote the migration, proliferation, and survival of CLL cells.30 Therefore, characterizing proteomics changes of CLL cells post stimulation of BCR can perhaps provide new insights into the contribution of BCR-signaling to CLL progression and may discover novel targets for CLL therapy. Based on this rational, Kashuba et al applied 2DE and mass spectrometry on 3 CLL samples to study proteomics alterations induced by stimulation of BCR.31 The study reported differential expression of 16 proteins, of which low molecular weight kininogen (LMWK) was up-regulated in all 3 samples, indicating a role of this protein in BCR signaling of CLL cells. Low molecular weight kininogen is an isoform of kininogen (plasma kallikrein; KLKB1), which is an important player of the kinin-kallikrein system that has been implicated in different biological processes including cell migration, proliferation and inflammation.32 The authors also reported a basal expression of LMWK in 71% of 51 CLL samples.31 In contrast, LMWK was not detectable in normal B-cells from healthy donors (n=4), suggesting an aberrant expression of LMWK in CLL cells.31 Consistent with these findings, Adamopoulos et al reported a dramatically increased expression of kininogen transcript in CLL samples relative to normal B-cells from healthy donors.33 These data provided evidence of the possible diagnostic usefulness of kininogen and its isoform LMWK to differentiate between CLL cells and normal B-cells. Furthermore, the preferential expression of kininogen and LMWK in CLL compared with normal B-cells and the up-regulation of LMWK post BCR stimulation indicated that these 2 proteins have roles in the pathology of CLL. This view was supported by the findings reported by Kashuba et al, where an increased expression of LMWK was associated with short overall survival of CLL patients (p=0.12).31 Overall, these findings encourage further investigations to examine the therapeutic potential of targeting LMWK to inhibit BCR-dependent progression of CLL.
The mutational status of IGHV significantly impacts on the responsiveness of BCR stimulation of CLL cells.4 At the cellular level, BCR-dependent proliferation was reported to be restricted to UM-CLL cells.34 Consistently, BCR activation was shown to induce much greater transcriptomics changes in UM-CLL in comparison with M-CLL.34 Differential proteome of BCR signaling in UM-CLL cells and M-CLL was sought by Perrot et al using 2-dimensional difference gel electrophoresis (2D-DIGE) and 1DLC connected with tandem mass spectrometry.35 The study was conducted on 3 UM-CLL samples (CD38+ and ZAP-70+) and 3 M-CLL samples (CD38-and ZAP-70-). In agreement with the notion that BCR-dependent cellular and molecular changes that promote CLL progression are limited to UM-CLL, dramatic alterations in the proteome of CLL cells post BCR-activation were recorded only for the UM-CLL samples. In fact, differential expression after BCR-activation was shown for 25 proteins in the UM-CLL samples as opposed to only 6 proteins in the M-CLL samples. Collectively, these proteins were implicated in cytoskeleton activity, cell growth, apoptosis, metabolism and signal transduction. At the protein level, these findings provided new insights into the variable response to BCR-stimulation in UM-CLL and M-CLL; and unveil a number of potential targets to inhibit BCR-dependent biological processes that favor the tumor progression. Of interest, programmed cell death protein 4 (PDCD4), which suppresses tumor growth and inhibits translation initiation,36 and UV excision repair protein (RAD23B, also known as hHR23B), which is implicated in the repair of DNA damages and function of p53,37 were down-regulated following BCR-activation in UM-CLL samples. In line with these findings, a transcriptomics study reported a striking up-regulation of genes implicated in mRNA translation and down-regulation of PDCD4 following ligation of BCR.38 B-cell receptor signaling occurs in the microenvironment of the lymph node, where a diverse set of proliferation and survival signals are delivered to CLL cells,26 thus the reduced expression of PDCD4 seems essential to allow the translation of transcripts that drive the growth and survival of the malignant cells. Defective or less functioning p53, which is a major tumor-suppressor protein, promotes the expansion and survival of CLL cells and hence contributes significantly to adverse clinical outcomes of the disease.17 As a result, the reduced expression of RAD23B post BCR-activation reinforces the important roles of aberrant p53 in the pathogenesis and prognosis of CLL. In contrast to the under-expressed proteins, heterogeneous nuclear ribonucleoprotein K (HNRNPK) and lymphocyte-specific protein-1 (LSP-1) were shown to be up-regulated in the UM-CLL samples upon BCR-activation. HNRNPK plays roles in pre-mRNA processing and BCR signaling; and drives the progression of lymphoma and leukemia.39-41 Lymphocyte-specific protein-1, which is also known as 47 kDa actin-binding protein, is subjected to phosphorylation by protein kinase C and was shown to be over-expressed in CLL cells as opposed to normal B-cells and to play roles in the survival of CLL cells.35,42,43 These findings give additional clues about BCR-dependent growth and survival of CLL cells, which can be utilized for the treatment of the disease.
Proteomics revelations of driver mutations in CLL
Abnormalities of translocation-associated notch protein TAN-1 (NOTCH1) pathway augment the survival of CLL cells and contribute to undesired clinical outcomes of the disease.4 In fact, various studies associated mutated NOTCH1 with early therapy and short overall survival in CLL patients.44-46 Functional analysis of the impact of NOTCH1 mutations on CLL cells reported significant activation of NOTCH1 pathway leading to an enhanced survival of CLL cells.47 Interestingly, the impact of NOTCH1 mutations appeared to be microenvironment-dependent.47 Utilizing antibody microarray-based proteomics, Díez et al compared the expression of 224 proteins in CLL samples (n=8) with or without NOTCH1 mutations.48 The study reported decreased expression of telomeric repeat-binding factor 1 (Trf-1), which is implicated in negative regulation of DNA duplication and promotion of apoptosis,49,50 protein kinase C gamma type (PKC g), which plays roles in the slow progression of CLL seen in good prognosis of the disease,43 and cyclin-dependent kinase inhibitor 2A (also know as p14arf), which inhibits cell cycle progression and incudes apoptosis.51,52 These findings, suggest an explanation of the known role of mutated NOTCH1 on the progression of CLL. This view was further supported by the findings obtained from the compression between mutated NOTC1 CLL samples (n=4) and normal B-cells samples (n=63), where proliferation of CLL cells appeared to be enhanced by decreasing the expression of p21 CDK inhibitor and increasing the expression of cyclin-dependent kinase 4 (Cdk4) and 6.
TP53 encodes for an important tumor suppresser protein know as p53.53 Loss or mutation of TP53 were associated with poor prognosis of CLL.4 Interestingly, Díez et al reported low-expression of PKC-related proteins in CLL cells harboring alterations in TP50.48 Given the involvement of PKC proteins in the slow progression of CLL,43 their decreased expression may explain the aggressive form of the disease associated with mutated p53.
Specific proteomics signature of CLL cells compared with normal B-cells
The identification of differentially expressed proteins in CLL cells compared with normal B-cells has the potential to describe novel mechanisms that underlie the malignant transformation into CLL and to reveal candidates with the potential to be used as targets for therapy and/or biomarkers for diagnosis. Johnston et al studied the proteomes of CLL samples (n=14) and normal B-cells from healthy donors (n =3) using tandem mass tag (TMT) with 2DLC and tandem mass spectrometry.54 A large coverage of proteomics was reported by this study, where more than 8100 proteins were identified in the 2 groups of samples, from which 544 proteins were up-regulated and 592 proteins were down-regulated in the malignant cells. The authors reported a proteomics signature specific to CLL irrespective of the disease subtypes as defined by mutational status of IGHV, expression of ZAP-70 and CD38 and chromosomal abnormalities. Strikingly, 95 of the over-expressed proteins in CLL cells were linked to mRNA processing and splicing, indicating that defects in such biological processes have major roles in the pathogenesis of the disease. Among the 95 proteins are different types of splicing factors (SFs), pre-mRNA splicing factors (PRPFs), small nuclear ribonucleoproteins (SNRNPs) and heterogeneous nuclear ribonucleoproteins (HNRNPs). Another piece of evidence of the involvement of aberrant mRNA splicing in the pathogenesis of CLL came from genomics investigations that found mutations of splicing factor 3B1 gene (SF3B1) in 4-5% of CLL patients at diagnosis with an increased mutation rate (17-18%) during the progression of the disease.55 Furthermore, mutated SF3B1 was reported to predict faster progression of CLL combined with early therapy and short overall survival.46 In accord with the findings of Johnston et al,54 SF3B1 was previously reported to be hypomethylated and over-expressed in CLL cells relative to normal B-cells.55,56 Although lesions of SF3B1 is a determinant of the progression and prognosis of CLL, the altered mRNA splicing reported by Johnston et al in CLL cells was independent of SF3B1 mutations,54 indicating that defective spliceosome is a hallmark of CLL cells (irrespective of the disease prognosis) compared with normal B-cells. Novel membrane-associated proteins including cytoskeleton-associated protein 4 (CKAP4), polymeric immunoglobulin receptor (PIGR), transmembrane and coiled-coil domain protein 3 (TMCC3) and CD75; and others that are involved in BCR signaling like lymphocyte transmembrane adapter 1 (LAX1), C-type lectin domain family 17 member A (CLEC17A) and plasma membrane calcium-transporting ATPase 4 (ATP2B4); were also reported with elevated expression in the CLL samples. Given the preferential association of these proteins with CLL, they may be good candidates as targets for immunotherapy and as biomarkers for diagnosis. Moreover, these data elucidate an aberrant BCR signaling of CLL cells as demonstrated by the increased expression of LAX1, CLEC17A and ATP2B4, agreeing with previous studies where peculiar BCR signaling was described in CLL.4,30 For example, ZAP-70, which is undetectable in normal B-cells, was found to be expressed in CLL samples from a subset of patients and to serve as a BCR signal transducer.57 Using existing inhibitors/drugs knowledge via ingenuity pathway analysis (IPA), Johnston et al proposed a number of inhibitors of the highly expressed proteins in CLL as a mean of therapy.54 For instance, G2 checkpoint kinase WEE1 could be targeted by the inhibitor MK1775 to repress cell cycle progression. In addition, entinostat was identified as an inhibitor of histone deacetylase 1 and 3 to induce apoptosis in the tumor cells. Collectively, this study provided novel clues about the pathogenesis of CLL and proposed therapeutic targets and diagnostic biomarkers of the disease.
From surface proteome to discovery of novel proteins and potential diagnostic markers of CLL
Searching for altered expression of cellular surface proteins is an attractive strategy for the discovery of biomarkers. Miguet et al employed 1DE with 1DLC and tandem mass spectrometry to characterize the proteome content of plasma-membrane derived from 3 types of hematological malignant cells; CLL cells, small cell lymphoma (SLL) cells and mental cell lymphoma (MCL) cells.58 The study was conducted on one sample from each type of tumor and reported a total proteome containing 597 proteins, of which 33 proteins displayed potential discriminatory value between the 3 illnesses. Subsequent validation analyses of the diagnostic potential of one of these proteins in 158 patients using flow-cytometry confirmed a significance of CD148 over-expression as a diagnostic biomarker that indicates MCL and exclude CLL and SLL. A later study reached a similar conclusion, where increased expression of CD148 and a high CD148/CD200 ratio clearly differentiated between MCL and CLL in a cohort that consisted of 374 patients.59 Boyd et al used 1DE with mass spectrometry and 1DE followed by 1DLC and tandem mass spectrometry to study the proteome of CLL plasma membrane.60 The authors reported 500 proteins, of which nearly 50% were membrane-associated proteins. Intriguingly, the study discovered novel proteins, such as MIG2B (also known as fermitin family homolog 3; FERMT3) and B-cell novel protein1 (BCNP1; also known as FAM129C). MIG2B was later found to have a major role in the adhesion of hematological cells, and to denote short overall survival of patients with multiple myeloma, which is another type of B-cell cancer.61,62 Recently, BCNP1 was also shown to be altered in various kinds of cancer; and that defects of BCNP1 was associated with molecular alterations of TP53, which encode for the tumor suppressor protein p53.63 Further analyses of the MIG2B and BCNP1 at the level of transcript revealed an association between these 2 transcripts and B-cell malignancies including CLL.60 The proposed importance of MIG2B and BCNP1 in cancer and their association with B-cell malignancies call for additional investigation to determine their roles in CLL.
From proteomics to discovery of potential prognostic markers of CLL
Although several different biomarkers of CLL are well established and widely implemented to predict the clinical course of CLL, the prognostication of CLL remains challenging.64 Therefore, the search for novel prognostic markers of CLL is still needed. Voss et al employed 2DE coupled with mass spectrometry to study the proteomes of CLL samples from 24 patients with variable prognoses.65 The study identified 16 proteins with differential expression that predicted chromosomal aberrations and overall survival in CLL patients. Notably, reduced expression of enzymes that were implicated in the detoxification of reactive oxygen species, such as thioredoxin peroxidase 2, glutathione S-transferase P (GST) and protein disulfide isomerase precursor (PDI), were reported to significantly identify patients with poor prognosis as defined by the 17p abnormality and/or short overall survival. The increased expression of these 3 proteins was previously implicated in drug-resistance or inhibition of apoptosis.66-68 Therefore, it is not clear how the low levels of these proteins contribute to adverse prognosis of CLL. On the other hand, heat shock protein 27 (HSP27) was reported as a marker of poor prognosis that was also associated with 17p and 11q deletions.65 HSP27 protects CLL cells from apoptosis and activates nuclear factor NF-kB pathway, providing an explanation of its ability to identify patients with an undesired prognosis.69,70 These findings support the view that poor prognosis of CLL is characterized by defective apoptosis and active NF-kB which lead to proliferation and survival of the cancer cells.4 Taking together, the findings shown by Voss et al provided a group of proteins with prognostic potential that merit further validation to confirm their prognostic significance of CLL.65
In our previous published work,71,72 we used 2DLC and tandem mass spectrometry with and without iTRAQ to conduct qualitative and quantitative proteomics analyses on CLL cells from 12 patients with different prognoses. We also integrated our CLL proteome with previously published transcriptomes of CLL cells and normal B-cells. The study reported a CLL proteome consisting of 728 proteins, of which 63 were found to be CLL-related proteins. Functional enrichment analyses showed that these proteins are implicated in activation of the NF-kB, gene expression/mRNA processing, nucleosome assembly, signal transduction and apoptosis. Interestingly, these findings agree with previous reports, where CLL was shown with a dysfunctional apoptosis system, aberrant mRNA processing and active NF-kB pathway.73,74 The relevance of a subset of these proteins to CLL was validated in an additional CLL cohort; and 4 proteins were reported to predict poor prognosis of CLL and to provide an explanation, at least partially, of the aggressive form of the cancer. For example, increased expression of the onco-protein T-cell leukemia/lymphoma protein A1 (TCL-1), which causes a CLL-like disease when over-expressed in mice, activates NF-kB and enhanced the response of BCR stimulation,75-77 was reported to predict an early therapy and to be significantly associated with other poor prognostic markers of CLL, like high expression of CD38 and ZAP-70 as well as un-mutated IGHV. In addition, elevated expression of S100A8 protein, which activates MAP kinase and NF-kB; and promotes the proliferation and metastasis of malignant cells,78,79 was found to identify a group of patients with rapid progression of the disease and an early treatment. Up-regulated expression of thyroid hormone receptor-associated protein 3 (Trap150 also known as TR150), which plays roles in the transcription activation and mRNA splicing of different genes including the cell cycle gene cyclin D1 that was implemented in the proliferation of CLL cells,80,81 was informative of an early need for therapy in CLL patients. In addition to its prognostic potential, the integration of our CLL proteome with the transcriptomes of CLL cells and normal B-cells showed a possible diagnostic usefulness of TR150 in CLL as evidenced by its specific expression in CLL cells as opposed to normal B-cells. This piece of information agrees with the view reported by Johnston et al that over-expression of proteins involved in mRNA processing and splicing is a feature of CLL in contrast to normal B-cells.54 Reduced expression of myosin-9, which is required for detachment of lymphocytes from adhesive molecules in the microenvironment of the lymph nodes and the egress of lymphocytes from lymph node to peripheral blood,82 was reported in our study to predict a progressive form of CLL and to associate with a high-expression of ZAP-70. Interestingly, this finding supports the data shown by Eagle et al, where the aggressive nature of UM-CLL was attributed to high adhesion and longer retention of the malignant cells in the lymph nodes.21 Collectively, this study sheds some light into the molecular pathology of CLL and reported a number of proteins with the potential to serve as prognostic markers of the disease.
In conclusion, in the current review the outstanding results of published CLL proteomics studies were examined and discussed. A summary of these findings is shown in Table 1. Overall, these findings painted a clearer picture of the molecular pathology of CLL and highlighted proteins with prognostic and/or therapeutic promise. Given the complex nature of the proteomic approach, limited numbers of CLL samples were used by the investigative studies reviewed here. Therefore, further validations using large patient cohorts are required to affirm the usefulness of the proteins of interest as biomarkers and/or therapeutic targets of CLL. It is important for the confirmatory studies to focus on proteins that are likely to provide additional improvements to the current prognosis and/or therapy of the disease. Additional targets for the validation studies are the proteins that probably serve as surrogate markers for the well-established prognostic markers that need advanced technology and high-level of expertise, such as the mutational status of IGHV.
Summary of the major findings of the chronic lymphocytic leukemia (CLL) proteomics studies that were discussed in the present review.
Acknowledgment
The authors would like to thank the Deanship of Scientific Research at Majmaah University in Saudi Arabia for supporting this study under a project number [1440-58]. The authors also would like to thank the American Manuscript Editors (www.americanmanuscripteditors.com) for English language editing. Figure 1 was drawn by the authors using BioRender application.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company. This study was funded by the Deanship of Scientific Research, Majmaah University, Majmaah, Saudi Arabia Gran number 1440-58.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.