


Abstract
Hyperphosphatemia is a major cause of morbidity and mortality in patients with chronic kidney disease. The association between hyperphosphatemia and increased risk of death from cardiovascular disease/vascular calcification has been well established for a long time. This review explores the new aspects of pathogenesis of vascular calcification, as demonstrated by recent advances showing a recognized regulating role of phosphorus in vascular smooth muscle cell calcification. This novel mechanism may help in finding a new pharmacological therapy to reduce, or prevent blood vessel calcification. Furthermore, recent experimental and clinical studies involved in the treatment of hyperphosphatemia are reviewed in this article.
Inorganic phosphorus is an intracellular anion. It plays an essential role in cell functions, including production of energy, membrane transport, and signal transduction.1 Klotho-fibroblast growth factor (FGF-23) system maintains phosphate (pi) hemostasis.2 Klotho works as a receptor for the phosphaturic hormone FGF-23. Phosphate retention represents a critical feature of pathophysiology of chronic kidney disease (CKD). In the community, elevated serum phosphate levels are associated with increased risk of cardiovascular disease (CVD) in individuals without history of CVD or CKD as shown by Dhingra et al.3 Even high normal blood phosphate levels in healthy normal individuals were associated with increased coronary artery calcium content with subsequent risk of CVD.4
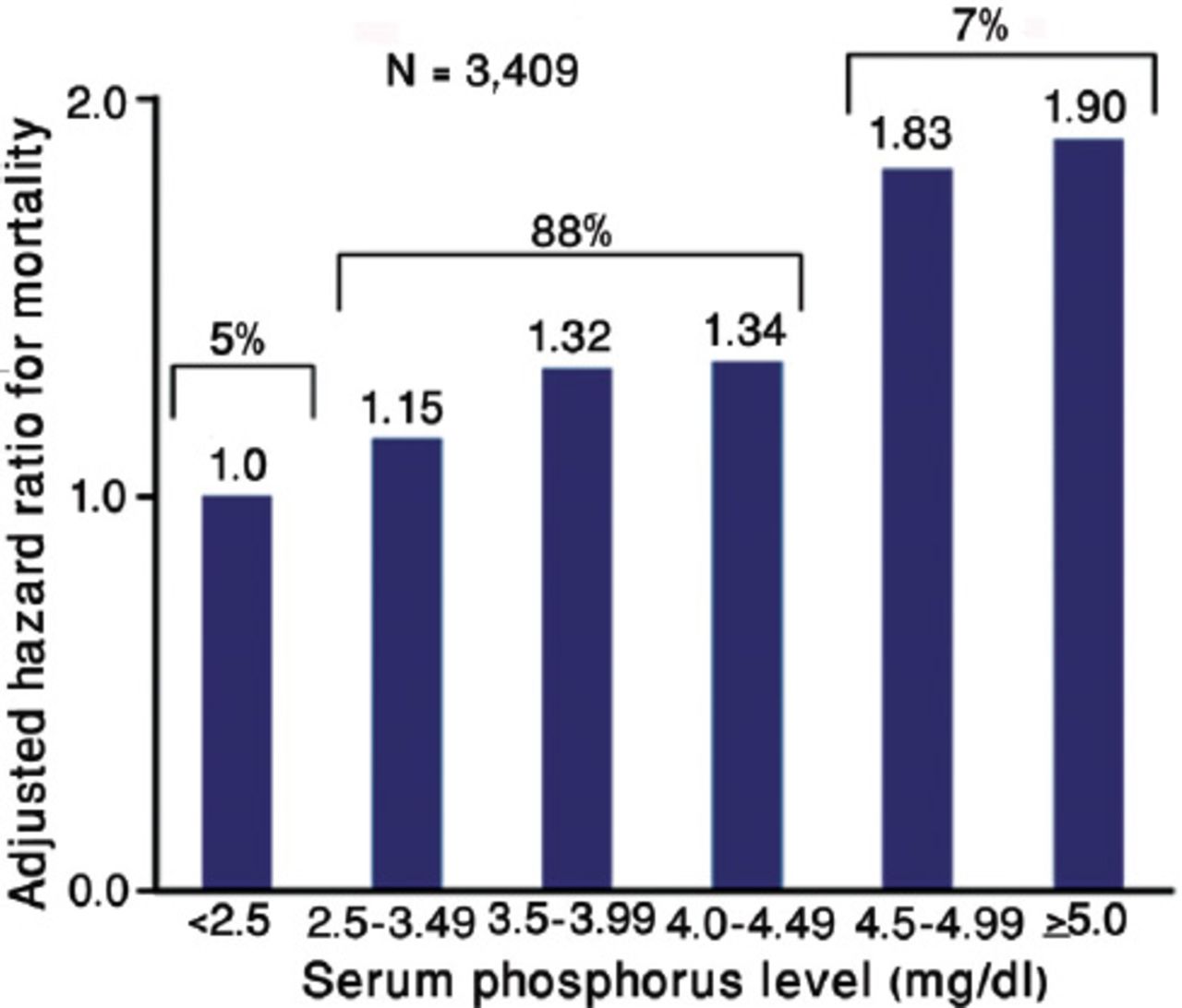
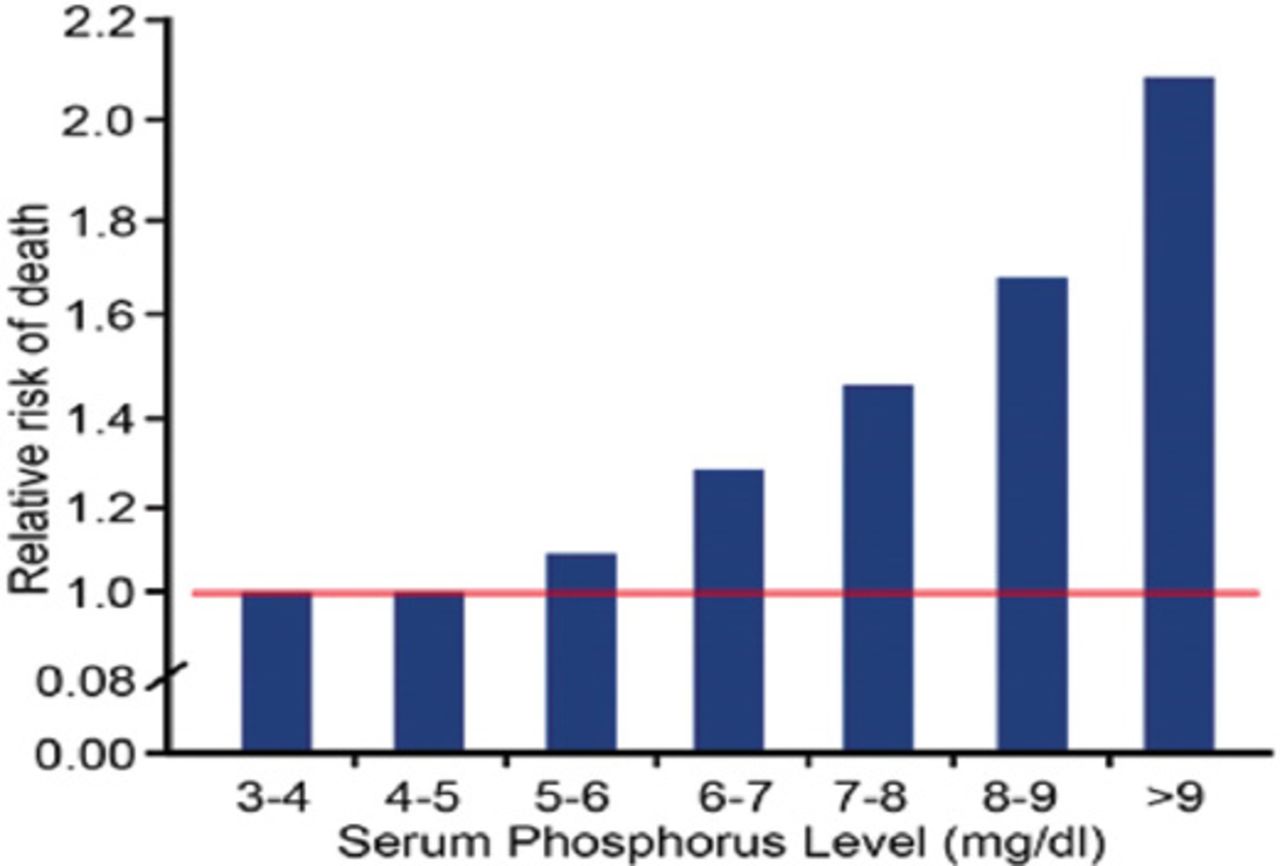
Hyperphosphatemia is independently associated with high mortality risk in CKD population, whether they are on dialysis, or not.5,6 Figure 1 shows the view of Kestenbaum et al,5 who noticed that serum phosphate levels of more than 3.5 mg/dL (1.13 mmol/L) were associated with significantly increased risk for death in CKD patients who were not on dialysis (creatinine clearance >50.4 - 39.5 ml/min). Earlier, a national study in the USA showed a 27% increase in relative risk (RR) of death, associated with serum phosphorus >6.5 mg/dL among people who received chronic dialysis.6 Then, Block et al6 again reviewed the database of >40,000 hemodialysis patients to find a strong association between higher levels of serum phosphorus (>5 mg/dL), and increased risk of death (Figure 2). More recently, Calo et al7 in their study on sevelamer-resistant dialysis patients found a relationship between the reduction of serum phosphate induced by chitosan-loaded chewing gum and C-reactive protein reduction, which support a proinflammatory role of hyperphosphatemia “per se”.

Mortality risk increases with serum phosphorus in patients with chronic kidney disease-stage 3 not on dialysis. Republished with permission from the American Society of Nephrology. Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol 2005; 16: 520-528.

Mortality risk increases with increased serum phosphorous in dialysis patients. Republished with permission from the American Society of Nephrology. Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15: 2208-2218.
The high mortality rate in CKD patients is related to CVD, which in fact, is responsible for more than 50% of deaths in patients with end stage renal disease (ESRD).8 Sudden cardiac death, heart failure, ischemic heart disease, and peripheral arterial disease are the primary causes of cardiovascular mortality.8-11 These cardiovascular events are strongly correlated with vascular calcification; a pathology which is induced and promoted by hyperphosphatemia, elevated calcium x phosphorus (Ca x P) product, and increased calcium load (diet, dialysate).6 Over the past 2 decades, several studies have shown the clinical significance of vascular calcification in CKD. These studies demonstrated that arterial calcification is strongly associated with coronary ischemic disease in uremic patients.8,12 Intimal calcification is classically associated with advanced stages of atherosclerosis and subsequent ischemic heart disease, while calcification of media is most likely dependent upon the abnormalities of mineral metabolism typically found in CKD patients with mineral and bone disorder (MBD). Medial arterial calcification leads to increased arterial wall stiffness, and increased pulse pressure resulting in the development of cardiomyopathy, arrhythmia, and sudden cardiac death.13 However, these 2 types of vascular calcification (intimal and medial) can co-exist in the same patient and the same vessel.14 In addition, calcification of cardiac valves and myocardium is also increased in dialysis patients leading to increased morbidity and mortality.10 Finally, vascular medial calcification of small arterioles is responsible for calciphylaxis, a syndrome of ischemic necrosis of the skin associated with extremely high mortality rates in uremic patients.8,12 This review aims to provide a comprehensive understanding of the pathogenesis of vascular calcification though exploring the new aspects in this regard, as demonstrated by recent advances showing a recognized regulating role of phosphorous in calcification of vascular smooth muscle cells (VSMC).
Pathogenesis
The hallmark of vascular calcification is the deposition of calcium phosphate in the form of hydroxyapatite in arteries, myocardium, and cardiac valves.8 Calcification of blood vessels was, until recently, considered to be a passive and degenerative process.15 However, after being extensively researched, it is currently recognized as an active cell-regulated process, controlled, and directed by calcification inhibitors and promoters.16,17 An imbalance of these factors, results in a phenotypic transformation of VSMC. These cells express bone-related genes, which can by stimulated by promoters, switching these cells from contractile cell type to a calcifying cell type to act as osteoblast/chondrocyte-like cells that secrete osteogenic proteins, such as osteocalcin, sialoprotein, alkaline phosphatase, and so forth.18,19 In addition to phenotypic change, death of VSMC by apoptosis can provide apoptotic bodies and necrotic debris to serve as nucleation sites for hydroxyapatite deposition.20 Loss of calcification inhibitors, such as pyrophosphate and matrix gla protein (MGP) results in spontaneous vascular calcification and increased mortality.8 Fetuin-A (a2-HS-glycoprotein) is one of the more powerful circulating inhibitors of hydroxyapatite formation; it is a member of cysteine protease inhibitors’ superfamily, synthesized in the liver, and found in high concentration in mammalian serum.21 Fetuin-A reacts as a negative acute phase protein in acute and chronic inflammatory states.
Several studies22-25 showed that a lower serum Fetuin-A concentration was associated with increased all cause and cardiovascular mortality in dialysis patients. From the opposite side; hyperphosphatemia, high calcium load, and hyperparathyroidism are the main inducer factors in the process of vascular calcification (Table 1). In a recent study, Adiney et al26 evaluated 439 patients with CKD stage 3 or 4, and no history of CVD. They found that higher serum phosphate concentrations were significantly associated with higher incidence of vascular calcification. For each one mg/dL increment in serum phosphorus, there was increased calcification by 21% (p=0.002) in coronary artery, and 33% (p=0.001) in thoracic aorta.
Inductors and inhibitors of vascular calcification.
Effect of hyperphosphatemia
Jono et al,15 examined the effect of phosphorus (pi) on calcification of human smooth muscle cells (HSMC) cultures in an in vitro model. The results showed that calcium deposition dramatically increased in media containing pi of 2 mmol/L compared with media of pi 1.4 mol/L (on day 9, calcified HSMC cultures versus uncalcified control: 210 versus 15 mcg/mg protein, p<0.01) This indicates that cell cultures are susceptible to calcification when cultured in media containing pi levels resembling those usually found in patients with hyperphosphatemia.15 These results, along with observations from other studies6,8 support the hypothesis that elevated phosphorus directly induce VSMC to increase phosphorus uptake via a type III sodium dependent phosphate cotransporter (NPC [pit-I]) resulting in mineralization and phenotypic modulation of VSMC through increasing expression of cbfa-I, a bone specific transcription factor with subsequent elaboration of a matrix that contains osteopontin and osteocalcin, which are major non-collagenous proteins found in the bone matrix.15 Phosphorous induced mineralization was inhibited by phosphonoformic acid,15 and pit-1 knockdown cells cultured in a medium with high levels of phosphate showed reduced phosphate uptake, less calcification, and decreased expression of osteogenic factors (cbfa-1/Runx2) in comparison with control cells.27 More recently, Zao et al28 demonstrated a clear link between phosphate induced calcification and oxidative stress of mitochondria, activation of nuclear factor-κB, and subsequent increased expression of osteogenic factors resulting in vascular mineralization. This novel mechanism of link and regulation between phosphorous, mitochondrial oxidative stress, and nuclear factor-κB might help in finding antioxidants, as a new therapeutic agents for vascular calcification.29
Effects of hypercalcemia
Similar to hyperphosphatemia, high serum calcium has also been associated with vascular calcification and increased mortality in patients with CKD.5,6 In dialysis patients, exposure to dialysate solution containing high calcium concentration can lead to increased serum calcium levels.30,31 The use of vitamin D enhances calcium absorption from the gut. Also, chronic use of calcium-based phosphate binders can induce calcium load in CKD patients leading to more vascular calcification.32
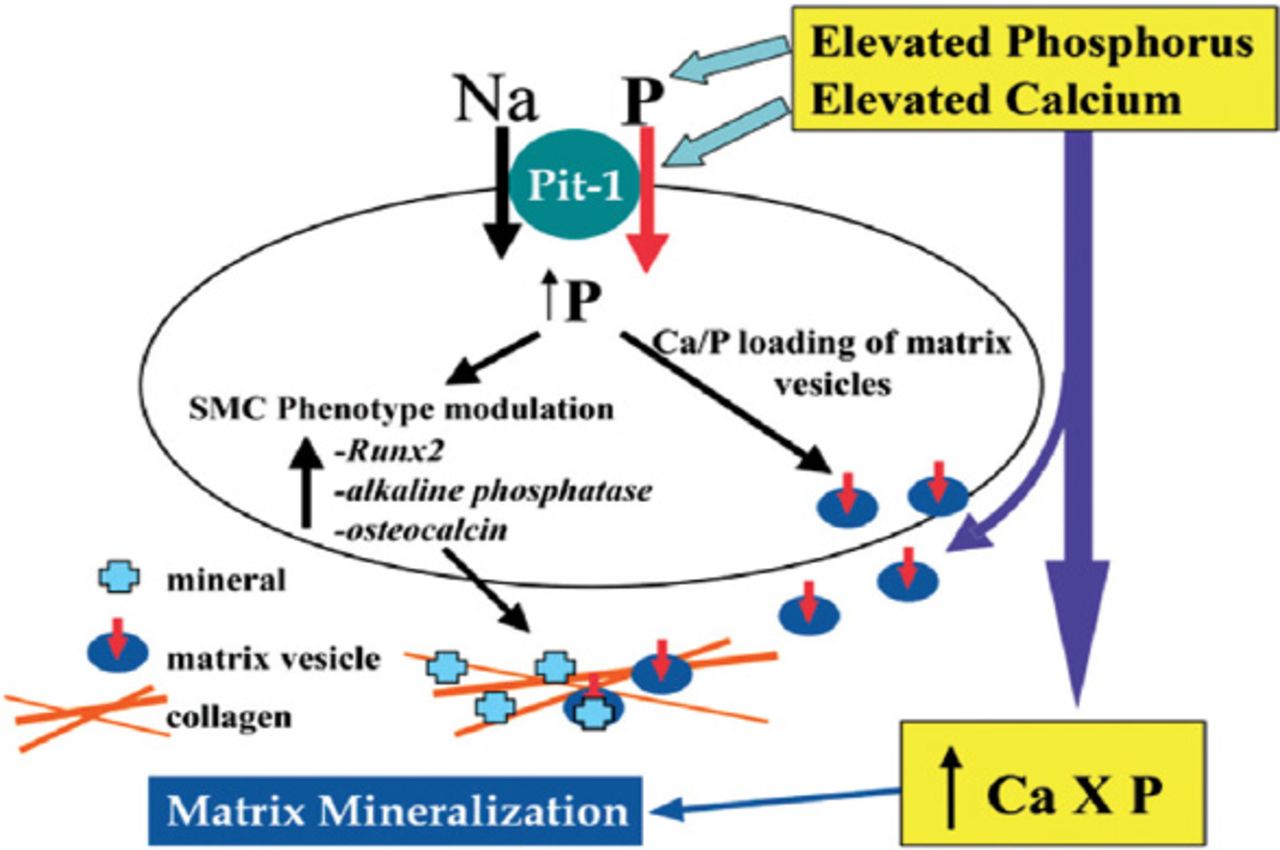
Yang et al12 examined the effect of increasing calcium concentrations on HSMC calcification in vitro. Results proved that increasing calcium to levels observed in hypercalcemic patients increased mineralization of VSMC cultures under normal phosphorus conditions. Furthermore, elevated levels of both calcium and phosphorus in the media led to accelerated mineralization. Again, this study showed that calcium induced mineralization was also suppressed by phosphonoformic acid (PFA) indicating the role of sodium dependent cotransporter in the mechanism of mineral deposition. Thus, authors concluded that hypercalcemia augments phosphorus uptake by NPC in VSMC, thereby leading to enhanced apatite nucleation in the extracellular matrix, and to phenotypic modulation of VSMC to act as calcifying cells with osteoblast/chondrocyte-like features (Figure 3).

Proposed model for the effects of elevated calcium (Ca) and phosphorus (P) on vascular smooth muscle cell (SMC) matrix mineralization. Republished with permission from the American Society of Nephrology. Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol 2004; 15: 2959-2964. Na - sodium
Effects of high levels of parathyroid hormone (PTH)
Recent studies showed high mortality rates in dialysis patients with hyperparathyroidism.33,34 In fact, it is very difficult clinically to evaluate the role of isolated PTH in CKD patients as hyperphosphatemia is typically accompanied by elevated PTH levels. Neves et al35 designed an experimental model to investigate the isolated effect of PTH on cardiovascular calcification in rats with chronic experimental uremia, as well as in those with normal renal function. The study showed that rats receiving high concentrations of PTH in continuous infusion developed massive aortic and coronary medial calcification, and this was observed in animals with severe hypocalcemia and hyperphosphatemia; in animals with hypercalcemia and normal phosphorus levels, and in animals with normal levels of calcium and phosphorus. In that study, authors demonstrated the relationship between high PTH levels and vascular calcification, which was described before in CKD patients.36 They also showed that high PTH levels14 led to vascular calcification even in the presence of low calcium or normal phosphorus levels, or even with normal renal function. The authors thought that vascular calcification described in those animals could be related to a direct effect of PTH on VSMC, and that continuous PTH infusion might lead to a decrease in calcification inhibitors, such as pyrophosphate, or MGP.35
Treatment
Treatment of hyperphosphatemia consists of 3 main ways (Table 2). Dietary phosphate restriction is the first step in the prevention and management of hyperphosphatemia. Protein restriction and avoidance of dairy products are the cornerstone of this regimen. Furthermore, extra phosphate load from food, which contain phosphate additives (cooked ham, roast breast turkey/chicken, and so forth), and high phosphate content of beverage creates a special concern because this extra amount of phosphorous is almost completely absorbed by the intestinal tract.37,38 Effective dialysis plays an essential role in decreasing high serum phosphate levels. This review will concentrate more on phosphate binders.
Modalities of treatment of hyperphosphatemia.
Calcium salts versus sevelamer
Calcium-containing phosphate binders can lead to hypercalcemia, and may exacerbate metastatic calcification.39 There is already a clear-cut evidence indicating not to use calcium-based agents in patients with positive calcium balance to avoid vascular calcification.40 Therefore, the use of non-calcium-based agents like sevelamer has become a commonly used therapy.
Experimental studies on phosphate binders showed a clear beneficial effect of these agents in suppressing ectopic calcification. For example, Katsumata et al36 examined the effectiveness of sevelamer in preventing ectopic calcification of soft tissues in adenine induced renal failure rats. Uremic rats demonstrated an increase in ectopic calcification of blood vessels, kidney, and stomach, as well as hyperphosphatemia after 3 weeks of adenine diet. When serum phosphorus level had significantly increased, the rats were freely fed a normal diet that contained one or 2% sevelamer for another 5 weeks. The results showed that compared with controls (n=10), sevelamer-treated animals (n=16) had reduced serum phosphorus, and calcification was clearly suppressed in their aorta media.
Clinical studies
Several clinical studies have been carried out on phosphate binders to evaluate their effects on hyperphosphatemia and vascular calcification. Giachelli8 in his review of vascular calcification mechanisms mentioned several studies, which show that high calcium intake increases calcification score, and the extent of arterial calcification increased with the use of calcium-based phosphate binders. Another study carried out by Block et al,32 compared the effects of sevelamer and calcium salts on coronary artery calcification in patients new to hemodialysis (HD). The results showed that the median absolute increase in Coronary Artery Calcification Score (CACS) at 18 months was 11-fold greater in the calcium group compared to the sevelamer-treated group. Again Block et al41 assessed all-cause mortality in 127 patients new to HD assigned to calcium-containing binders, or sevelamer after a median follow-up of 44 months from randomization; a total of 34 deaths occurred during the period of follow-up: 23 deaths in the calcium group, and 11 deaths in the sevelamer group (p=0.02, hazard ratio (HR): 3.1). The authors concluded that treatment with sevelamer was associated with a significant survival benefit, as compared with the use of calcium-containing phosphate binders. It seems that the effect of sevelamer on cardiovascular system is time-dependent. Suki et al,42 showed a clear improvement in survival among patients treated for more than 2 years with sevelamer, as compared with those treated with calcium salts (p=0.02, RR=0.66). This time related effect of sevelamer was further supported by Savica et al,43 who observed the effect of sevelamer on cardiovascular morbidity (CV hospitalization events and duration) for 3 years in 16 patients on maintenance dialysis. At the end of the study, there was a significant reduction in serum calcium and phosphorous. Also, there was a decrease in hospitalization rate (p<0.01) and hospitalization duration (p<0.001).
Lanthanum carbonate
Lanthanum carbonate, another non-calcium-based phosphate binder was assessed by Hutchison et al44,45 on patients who received either hemodialysis, or peritoneal dialysis treatment for ESRD. The results showed that lanthanum carbonate effectively reduced serum phosphate levels, and when used as a monotherapy, it provided effective phosphate control with a low tablet burden.
Nicotinamide
Takahashi et al46 showed in their clinical study on 65 hemodialysis patients that Nicotinamide, a metabolite of nicotinic acid (Vitamin B3) decreased serum phosphorus levels significantly from 6.9±1.5 mg/dL to 5.4±1.3 mg/dL during the 12-weeks of Nicotinamide treatment. Nicotinic acid is known to be an inhibitor of sodium dependent phosphate cotransporter in renal tubule and small intestine of rats.
Chitosan
Increased excretion of salivary phosphate has been reported in patients with CKD, and in hemodialysis patients’ independent from food intake.47,48 In an attempt to improve the treatment of hyperphosphatemia, Savica et al49 assessed the phosphate-binding capacity of the natural cationic polymer-chitosan as chewing gum (the 20 mg chitosan loaded chewing gum was manufactured as cold compressed 3-layer tablets). Thirteen hemodialysis patients with serum phosphate levels >6.0 mg/dL despite treatment with sevelamer hydrochloride chewed 20 mg of chitosan-loaded chewing gum twice daily during fasting (that is, between meals) for 2 weeks in addition to their prescribed phosphate-binding regimen. Salivary phosphate and serum phosphate significantly decreased during the first week with chewing. By the end of the second week, salivary phosphate decreased by 55% from baseline, and serum phosphate decreased 31% from baseline. Parathyroid hormone and serum calcium concentrations were not affected by the gum. These results suggest that adding salivary phosphate-binding to traditional phosphate binders could be a useful approach for improving treatment of hyperphosphatemia in dialysis patients.49,50
Iron salts
Iron salts are becoming a new phosphate binder still under investigation. Several studies suggest that ferric compounds can reduce intestinal phosphate uptake in uremic and non-uremic rats.51 So far, 2 compounds may enter the market in the near future; ferric citrate compound,52 and iron (III)-oxyhydroxide-based phosphate binder (PA21).53 Efficacy and short-term safety have almost been proven for both compounds, however, side effects and cost-effectiveness needs to be evaluated.
In conclusion, hyperphosphatemia is associated with increased risk of cardiovascular mortality in patients with CKD; this is mainly due to vascular calcification, which is currently considered as an active cell-regulated process. Hyperphosphatemia stimulate VSMC to convert from contractile cell type to an osteogenic cell type, through an increased intracellular phosphorus uptake via sodium dependent phosphate co-transporter (Pit-1), thus leading to vascular calcification. Aggressive management of hyperphosphatemia through dietary phosphate restriction, effective dialysis, and different kinds of phosphate binders is needed for optimal control of serum phosphate, in order to avoid calcification of blood vessels.
Related Articles
Kari JA, Eldesoky SM, Bagdadi OT. Vitamin D insufficiency and treatment with oral vitamin D3 in children with chronic kidney disease. Saudi Med J 2012; 33: 740-744.
Al-Sayyari AA, Shaheen FA. End stage chronic kidney disease in Saudi Arabia. A rapidly changing scene. Saudi Med J 2011; 32: 339-346.
Footnotes
Disclosure. This study was supported by the College of Medicine Research Center, Deanship of Scientific Research, King Saud University, Riyadh, Kingdom of Saudi Arabia.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.