


Abstract
Diffuse alveolar hemorrhage is an uncommon and often fatal condition in children that is characterized by distinct histopathological etiologies. Herein, we discuss the case of an 11-year-old girl who presented with acute worsening of hypoxia and left-sided chest pain. The patient had lung biopsy-proven idiopathic pulmonary capillaritis and was being treated with prednisolone every alternate day, azathioprine, and hydroxychloroquine. A contrast-computed tomography (CT) scan of the chest showed an acute left lower-lobe pulmonary embolism. Negative results were obtained on a test for thrombophilia. In children, pulmonary embolism with anti-neutrophil cytoplasmic antibody-negative idiopathic pulmonary capillaritis is a rare clinical condition. The exact cause of thrombus formation in this case is unknown; however, obesity, immobility, and chronic systemic corticosteroid therapy probably played a role.
Diffuse alveolar hemorrhage (DAH) is an uncommon condition caused by a variety of disorders.1,2 A DAH event can be life-threatening; therefore, timely intervention is critical. The DAH can mimic infectious and non-infectious disorders of the lower respiratory tract, such as chronic bacterial and fungal lung infections; pulmonary edema, chronic aspiration syndrome, and hypersensitivity pneumonitis. The diagnosis of DAH is often delayed due to the rarity of hemoptysis as a presenting symptom of DAH in children.1 Common respiratory symptoms in children with DAH include cough, dyspnea, and hypoxia. Radiographic abnormalities (alveolar or mixed alveolar/interstitial patterns) are also common; however, such findings are non-specific. The DAH can occur as part of a systemic disease or as an isolated lung pathology. Isolated DAH lung pathologies have recently been classified as DAH with and without capillaritis.1 Isolated capillaritis is characterized by inflammatory disruption of the capillary network, which leads to leakage of blood into the alveoli and interstitium.1,2 Idiopathic pulmonary capillaritis (IPC) is associated with high mortality and morbidity due to recurrent DAH, which is usually resistant to treatment. Autoimmune and vasculitic disorders can present with DAH caused by pulmonary capillaritis.1,3 Although, anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis is considered a hypercoagulable disorder associated with an increased risk of thromboembolic events,4 there have been no reports on the risk of thrombosis in children with ANCA-negative capillaritis. In this report, we have described a rare case of ANCA-negative IPC complicated by acute pulmonary embolism.
Case Report
An 11-year-old girl with recurrent DAH presented with worsening hypoxia and left-sided chest pain. The patient was first admitted at 8 years of age due to a sudden episode of pallor and jaundice (Table 1). A physical examination revealed that the child was well-nourished and had significant pallor and mild jaundice. Neither respiratory distress, nor organomegaly were observed. Verbal consent was obtained from the patient’s family and documented in her medical chart before the creation of this report.
Timeline from the first presentation to final diagnosis.
Clinical findings
At first hospital admission, the laboratory findings were as follows: hemoglobin (Hb) level 3.6 g/dL (reference range [RR], 11.5-15.5 g/dL), mean corpuscular volume 69.4 fL (RR, 77.0-95.0 fL), mean corpuscular hemoglobin level 19.6 pg (RR, 25.0-33.0 pg), and reticulocyte count 9.65% (reference level, <2%). Her white blood cell count and platelet count were within normal limits. A blood smear showed moderate anisopoikilocytosis with polychromasia. Her serum lactate dehydrogenase level was elevated to 887 IU/L (RR, 125-243 IU/L), and her haptoglobin level was normal (35 mg/dL; RR, 11-220 mg/dL). A direct Coombs test revealed negative results. Her serum ferritin level was 71 ng/mL (RR, 9-185 ng/mL), and her iron and transferrin values were normal. However, the transferrin saturation was borderline low. Extensive testing to determine the underlying cause of anemia revealed normal findings; she underwent an upper endoscopy and tests to detect anti-gliadin and tissue transglutaminase antibodies, which are indicators of celiac disease; the results were all negative. She received a blood transfusion due to symptomatic anemia, and oral iron supplement therapy was started given her borderline low transferrin saturation. Table 1 provides a timeline of the patient’s care.
Diagnostic assessment
Four months after the first hospital admission, she was admitted with fever, cough, and hypoxia. A nasopharyngeal swab was positive by real-time PCR for respiratory syncytial virus and influenza. A chest X-ray (CXR) revealed multifocal air-space disease consistent with pneumonia. Her Hb level was 9.6 g/dL. The patient received intravenous antibiotics and supplemental oxygen and was discharged on 1 L/min night-time oxygen due to frequent night-time hypoxia. Tests for primary immunodeficiency and human immunodeficiency virus were negative.
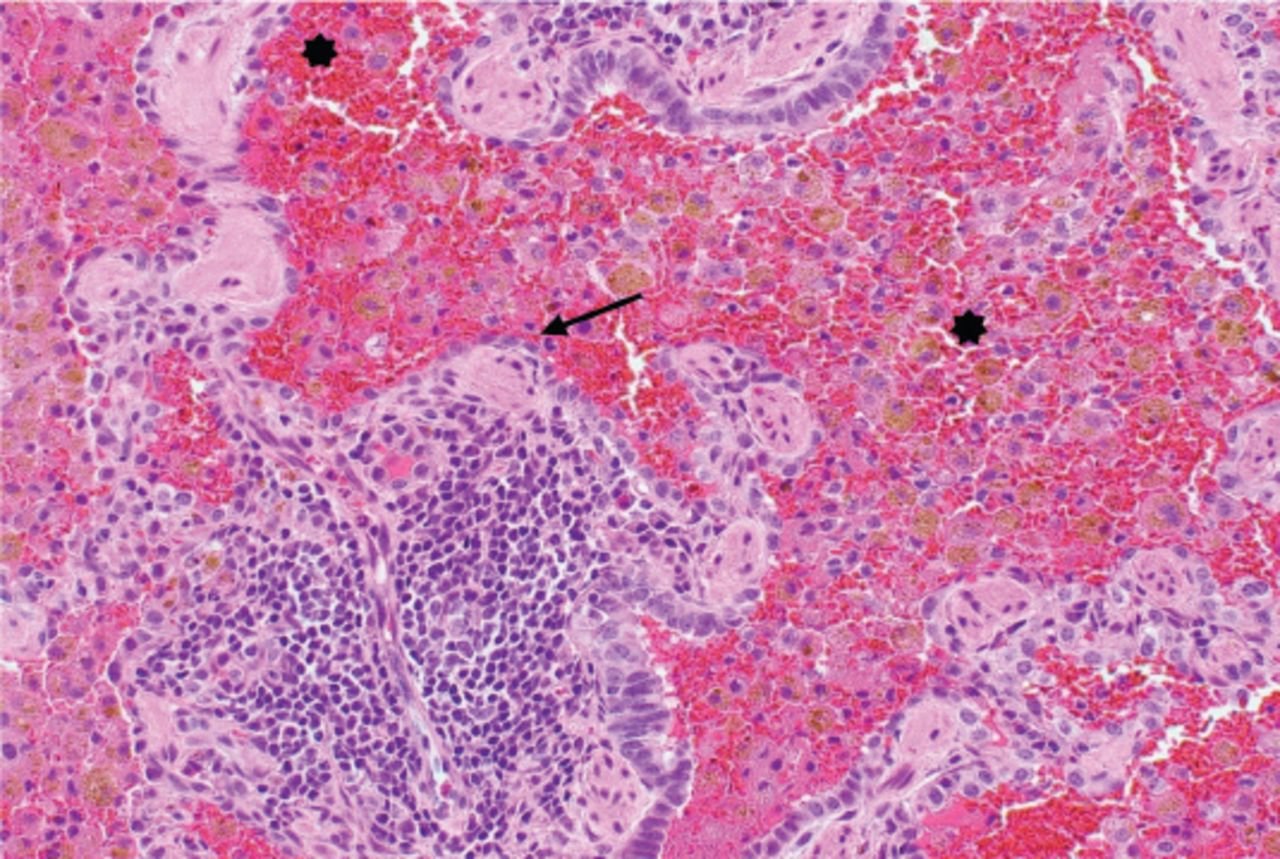
She continued to have frequent episodes of respiratory insufficiency and anemia with persistent CXR opacities involving the upper lobes. A CT scan of the chest showed diffuse airspace disease of both lungs that was most pronounced in the upper lobes. The chronic anemia, recurrent respiratory illnesses, and bilateral lung opacities suggested recurrent alveolar hemorrhages. Flexible bronchoscopy and open lung biopsy were performed. Bronchoalveolar lavage of the right middle lobe showed blood-stained fluid and yielded abundant hemosiderin-laden macrophages. A lung biopsy revealed a recent alveolar hemorrhage as well as signs of chronic changes most likely due to previous bleeding episodes. The signs of chronic changes included abundant hemosiderin-laden macrophages in the interstitium and alveolar spaces, and iron encrustation of vascular structures. No evidence of active vasculitis or capillaritis was observed (Figure 1). An autoantibody panel revealed negative results for anti-glomerular basement membrane antibodies, rheumatoid factors, ANCAs, anti-nuclear antibodies, proteinase-3 antibodies, anti-cardiolipin IgG antibodies, beta-2 glycoprotein antibodies, anti-Smith antibodies, and anti-dsDNA antibodies. These findings supported a diagnosis of idiopathic pulmonary hemosiderosis.

Numerous red blood cells in the alveolar space and abundant hemosiderin-laden macrophages in the interstitium and alveolar spaces (*). A lymphoid follicle is present in the center of the image (arrow).
Therapeutic intervention
The patient was admitted and treated with a methylprednisolone pulse dose for 3 days. Treatment was continued with a low dose of oral prednisolone. Under this course of treatment, frequent acute alveolar hemorrhages occurred; she was therefore transitioned to a regular steroid treatment with 10-20 mg of prednisolone every alternate day in addition to azathioprine and hydroxychloroquine.
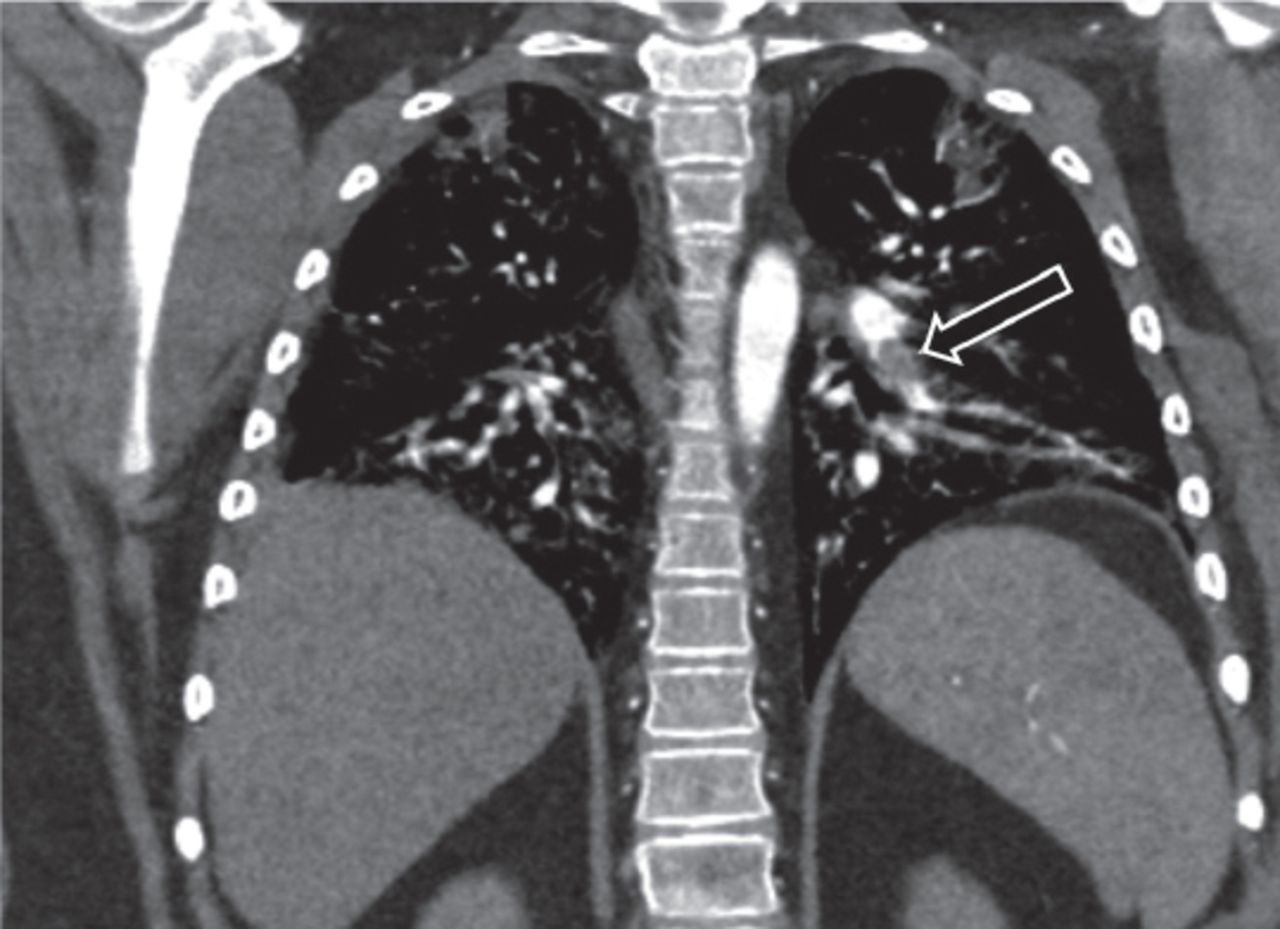
When the patient was 10 years of age, she presented with worsening hypoxia and left-sided chest pain. She had no fever, hemoptysis, leg pain, or swelling. An examination revealed tachypnea, retractions, and an oxygen saturation of 94% on nasal cannulation at 5 L/min. Chest auscultation revealed decreased breath sounds at the base of the lungs and inspiratory crackles. Her Hb level was 13.3 g/dL, and her hematocrit level was 39.8%. A CXR revealed bilateral reticular opacities that were markedly improved from those observed in the previous CXR, which was obtained prior to discharge, when she had the last episode of DAH 3 months before this presentation. A chest CT showed an acute left lower lobe pulmonary embolism and bilateral patchy airspace disease (Figure 2). A Doppler ultrasound of the lower limbs showed no evidence of deep vein thrombosis (data not shown). The results of a thrombophilia test to measure C and S proteins, antithrombin III, prothrombin gene mutations, and factor V Leiden were negative. The patient was started on low-molecular-weight heparin (enoxaparin sodium) at 1 mg/kg twice a day with frequent monitoring of anti-Xa levels which was later stopped after 6 months as per hematologist recommendations.

Left pulmonary artery embolism (black arrow).
Follow-up and outcome
The patient continued to have frequent alveolar hemorrhages despite prednisolone administration every other day as well as daily hydroxychloroquine and azathioprine administration. A lung biopsy sample was reviewed at another Childhood Interstitial Lung Diseases (chILD) Center, and neutrophilic infiltration of the alveoli and interstitium were observed. Therefore, a diagnosis of IPC was established, and induction therapy with rituximab and monthly intravenous immunoglobulin was started. Since August 2017, the patient has been stable on 2 L/min oxygen via nasal cannula with very mild episodes of alveolar hemorrhages. She received monthly rituximab for 3 months and is now receiving azathioprine, hydroxychloroquine, and monthly intravenous immunoglobulin therapy.
Discussion
This case reveals 2 clinical challenges of DAH due to pulmonary capillaritis in children: 1) it is rare and challenging to diagnose; and 2) if it is complicated by thromboembolism, treatment is difficult. The patient in the reported case was initially diagnosed with idiopathic pulmonary hemosiderosis and had received treatment for 2 years without any improvement. She had continued episodes of life-threatening hypoxic respiratory failure due to DAH despite compliance to treatment. Histopathological findings were frequently reviewed to confirm the diagnosis.
The exact cause of thrombus formation in ANCA-negative IPC is unknown; however, obesity, immobility, and chronic systemic corticosteroid therapy probably played a role in this patient’s condition. Thromboembolism has been documented in numerous types of small blood vessel vasculitis, including granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis, and microscopic polyangiitis.4
The IPC is a rare etiology of alveolar hemorrhage1,2 in childhood. It has 2 common presentations: severe life-threatening DAH and chronic anemia, the latter of which is often the result of iron deficiency and causes chronic persistent respiratory symptoms.5 Our patient experienced chronic progressive respiratory symptoms with frequent alveolar hemorrhages, mainly triggered by acute viral illness. The IPC is diagnosed by histopathological observation of erythrocytes leaking into the interstitium, fibrinoid necrosis of the capillary walls, and intra-alveolar septal capillary occlusion by fibrin thrombi.6 In acute intra-alveolar bleeding, erythrocytes fill the alveolar space and disrupt gas exchange. In contrast, in chronic bleeding, macrophages engulf erythrocytes, causing hemosiderin deposition.7 No known etiology clearly explains the alveolar and capillary network destruction and thromboembolic events that occur in this disease. Various pathways have been shown to be involved in the underlying pathobiological defects of thromboembolism in ANCA-associated vasculitic disorders.4 Interactions between activated neutrophils and endothelial cells lead to massive endothelial damage and thrombus formation4 In addition, electron microscopic studies have shown that immune complex-mediated processes may be active, even in the absence of immune deposition.4 Studies in adults have shown that anti-endothelial antibodies play a role in the pathogenesis of isolated ANCA-negative pulmonary capillaritis.4
Pulmonary thromboembolism was previously reported in a child with ANCA-positive pulmonary capillaritis.6 There are some reports of similar cases in adults;7 however, there are no other reports regarding children with ANCA-negative IPC who presented with an acute pulmonary embolism. Our patient had no evidence of systemic involvement, and all tests for connective tissue disorders yielded negative results. Immunosuppressive therapy is considered the main treatment for this condition and pulse steroids are often needed in case of acute life-threatening bleeding. However, in case of the occurrence of a thrombus, anticoagulants should also be administered to alleviate the risk of further thrombus formation.
In conclusion, treatment of a thrombus in patients with recurrent alveolar bleeding is challenging, and the condition may have life-threatening consequences. Anticoagulation therapy should be monitored closely and ceased in the presence of any early signs of acute pulmonary hemorrhage. For IPC patients, regular follow-up is crucial to allow early identification of new organ involvement. Further pathobiological studies are needed to clearly identify the exact pathology underlying this association and to propose treatment options.
Acknowledgment
The authors would like to thank the patient and her family. They would also like to thank Editage (http://www.editage.com) for English language editing. Special thanks to Dr. Mohammed Algathradi, pediatric radiologist, Department of Radiology, King Khalid University, Saudi Arabia, who read and reviewed the radiological pictures.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received January 17, 2019.
- Accepted May 2, 2019.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.