


Abstract
The methionyl-tRNA synthetase (MARS) mutation is a very rare cause of congenital pulmonary alveolar proteinosis. We report a 6-month-old boy born with symmetrical intrauterine growth retardation presented with unexplained persistent tachypnea and hypoxemia associated with severe failure to thrive, anemia, hypoalbuminemia and hepatomegaly. Detailed pulmonary investigations including computed tomography chest scan, bronchoscopy and bronchoalveolar lavage revealed pulmonary alveolar proteinosis. Whole exome sequencing identified a homozygous novel variant in the MARS gene, c.854T>C p.(Ile285Thr).
Pulmonary alveolar proteinosis (PAP) is a disorder resulting from the accumulation of lipoprotein substances within alveoli. Compared with adults, primary PAP in children is usually caused by inherited disorders, including surfactant dysfunction secondary to defects in several genes such as ABCA3, SFTPB, SFTPC, and NKX2-1.1,2 Additionally, surfactant clearance disorders associated with variants of the CSF2RA and CSF2RB genes may cause PAP.1,2
Methionyl-tRNA synthetase (MARS) is in the aminoacyl-tRNA synthetase (ARS) group of enzymes that are essential during protein biosynthesis.3,4 It was first reported by Van Meel et al,4 as a cause of interstitial lung disease in an infant with a compound heterozygous mutation in the MARS gene. Extrapulmonary manifestations of the MARS mutation include liver failure, intermittent lactic acidosis, aminoaciduria, hypothyroidism and transfusion-dependent anemia.4
The purpose of this study is to report a rare case of PAP caused by a mutation in the MARS gene.
Case Report
Patient’s information
A 6-month-old boy was referred to our tertiary pulmonary service because of unexplained persistent respiratory distress and failure to thrive since birth. He was delivered at 36 weeks of pregnancy, with a symmetrical intrauterine growth retardation (IUGR). Otherwise, the pregnancy was uneventful, and he was admitted to the neonatal intensive care unit for 5 days because of unexplained tachypnea and respiratory distress, which improved during the neonatal period but never completely resolved. He is the first baby born to a young couple who were not consanguineous but were from the same tribe.
Clinical findings
Presenting with shortness of breath, persistent cough and intermittent fever, our patient was treated in different district hospitals as a case of bronchiolitis, pneumonia, recurrent wheezing and possible asthma. He received multiple courses of antibiotics, nebulized bronchodilators and inhaled steroids with no response. Also, he was diagnosed with gastroesophageal reflux disease (GERD) based on a barium meal findings associated with shortness of breath and cough after feeding. However, he did not show an improvement following anti-reflux management and nasogastric feeding. Despite an optimum nutritional management with a high protein and calorie formula, he continued to have failure to thrive.
Upon arrival to our center at the age of 6 months, he had persistent respiratory distress in form of tachypnea, intercostal and subcostal recession together with an oxygen saturation of 78% in room air. His breath sounds were decreased on both sides with no additional sounds. He had no dysmorphic features and his head circumference, weight and height were all below the third centile. The rest of his clinical examination revealed an enlarged liver with a span of 10 cm and axial hypotonia with preserved reflexes.
Diagnostic assessment
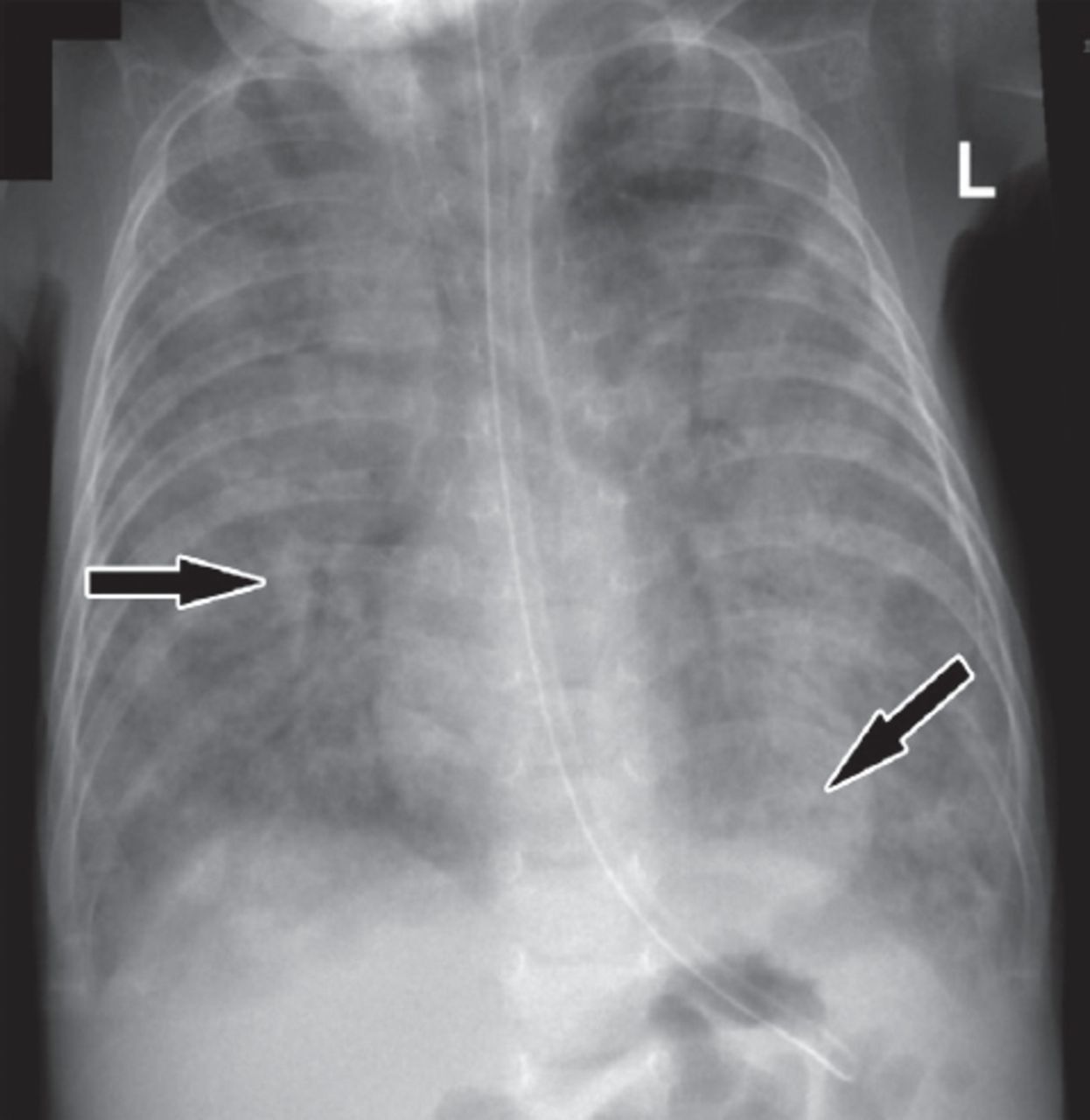
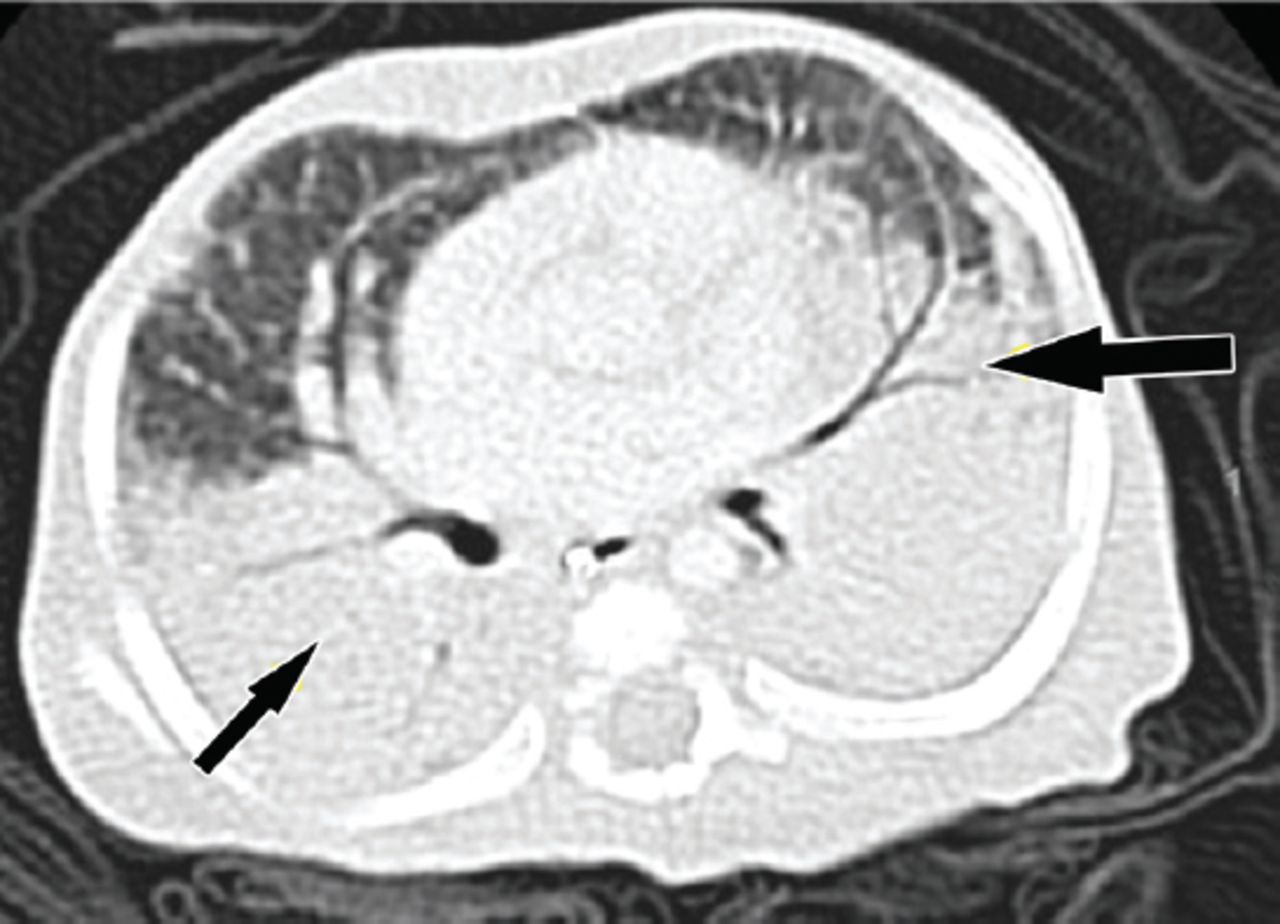
In an effort to unravel the underlying diagnosis of this patient, comprehensive investigations were planned by a multidisciplinary team from different pediatric subspecialties. The metabolic work up showed recurrent hypoglycemia; however, critical samples were normal. Therefore, hypoglycemia was attributed to his nutritional status. The endocrine work up revealed primary hypothyroidism with moderately elevated thyroid stimulating hormone and low thyroxine. His thyroid function normalized on thyroxine replacement therapy. An extensive hematology work-up revealed nutritional anemia and unexplained thrombocytopenia. Immunology investigations showed intact immunity with normal immunoglobulin and lymphocyte function. He had low albumin levels, but other liver function tests were normal and the urine examination showed no albuminuria. Thus, the low albumin was attributed to the nutritional status of the patient. He underwent extensive pulmonary investigations. His arterial blood gas was initially normal but later showed respiratory acidosis indicating emerging respiratory failure. His chest x-ray (Figure 1) showed diffuse alveolar disease, and his chest CT scan (Figure 2) showed bilateral basal infiltration and atelectasis with a positive Hounsfield unit rolling out intraparenchymal fat.

Chest x-ray showing a diffuse lung infiltrate, predominantly alveolar type, more prominent in the basal and peribronchial areas.

Chest CT scan showing extensive dependent atelectasis and airspace disease by air bronchogram suggestive of aspiration disease, Hounsfield unit (+30-65).
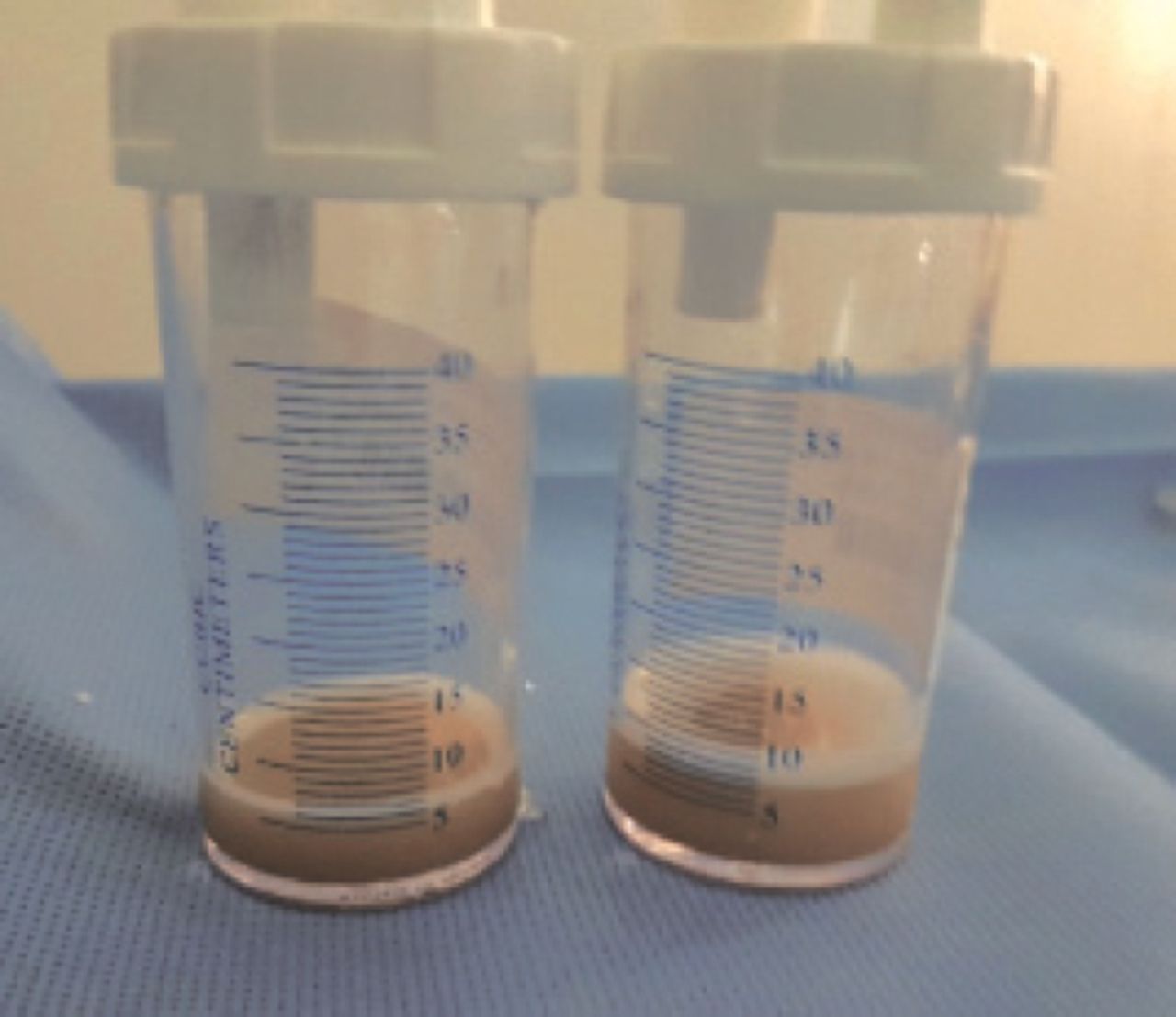
Bronchoscopy (Figure 3) showed a tracheal bronchus with otherwise normal structures. Bronchoalveolar lavage fluid samples were milky in color, and the cytology was negative for periodic acid-Schiff stain but positive for lipid-laden macrophages. The lavage cell count was high, 8442 C/L, with a neutrophilic predominance, and the cultures were negative. Despite the negative PAS, the bronchopulmonary lavage profile was suggestive of PAP.

Bronchoalveolar lavage fluid samples were thick, turbid and slightly mild with frothy secretions.
Therapeutic intervention
The family denied any history of feeding the patient high fat diet, nevertheless he was maintained on medium chain triglyceride (MCT) formula during his hospital stay prior to presentation.
Follow up and outcome
Despite intensive care support with conventional and high frequency ventilation, the clinical condition of the patient deteriorated, and he died due to respiratory failure. He was not stable for interventional therapeutic lavage.
Deoxyribonucleic acid (DNA) was extracted from the patient and his parents. Whole exome sequencing was performed using Illumina HiSeq (Illumina Inc., CA, USA).5
Whole exome sequencing revealed a novel homozygous variant of the MARS gene, c.854T>C p.(Ile285Thr). This variant was predicted to be damaging by PolyPhen, deleterious by Sift, and disease-causing by MutationTaster. The allele frequency was 0.000037 in ExAc and 0.00020 in 1000G. This variant segregated with the family as both parents were found to be heterozygous.
Discussion
In a child with suspected pulmonary alveolar proteinosis, the usual culprit is either a surfactant dysfunction disorder due to a defect in ABCA3, SFTPB, SFTPC, or NKX2-1, or a surfactant clearance disorder due to a defect in the CSF2RA or CSF2RB gene.1,2 Our patient had clinical suggestive signs of PAP; however, the gene panel study for surfactant dysfunction and clearance were normal. Therefore, whole exome sequencing was requested, which showed a homozygous variant of the MARS gene. Three in silico parameters predicted the variant to be disease causing, and the variant also showed a low allelic frequency (0.000210172341319882). Furthermore, a family segregation study was supportive of the pathogenicity of this variant. This variant is a non-conservative amino acid substitution that is likely to impact the secondary protein structure as these residues differ in polarity, charge, size or other properties. This substitution occurs at a position that is conserved across species. All these parameters make the variants more likely to be pathogenic.
Van Meel et al,4 were the first to report a MARS variant as a cause of interstitial lung disease in an infant who had a compound heterozygous mutation in this gene. Their patient also showed other features, including liver failure, intermittent lactic acidosis, aminoaciduria, hypothyroidism and transfusion-dependent anemia.4 By comparison, our patient had PAP in addition to some extrapulmonary manifestations, including severe failure to thrive, hypothyroidism, hypoalbuminemia, hepatomegaly, thrombocytopenia and anemia.
Enaud et al,6 reported 34 cases of PAP.6,7 Those patients showed radiological findings of a crazy paving pattern in 94% of cases and predominantly posterobasal consolidations in 76% of cases.6 Our patient showed a posterolateral consolidation that attributed to recurrent aspiration. Bronchoalveolar lavage fluid was obtained from 47% of cases, all of which were milky in appearance and positive for periodic acid-schiff (PAS) stain (Table 1).6 Other reported findings are liver disease in 91% of cases, anemia, and malnutrition with hypoalbuminemia.6The extrapulmonary manifestations of our patient were similar to those reported by Alice Hadchouel et al.5 Similarly, Sun et al,8 reported a family with 2 siblings with interstitial lung disease, mild liver dysfunction and failure to thrive. Whole exome sequencing revealed a compound heterozygous MARS variant.8
Comparison between our patient and reported cases from Réunion Island.
Timeline summary of the case.
In the Kingdom of Saudi Arabia, there have been reports of PAP due to surfactant clearance and surfactant dysfunction disorder.9,10 Nevertheless, this is the first report of PAP due to MARS mutation in the Arabian Gulf region
Methionyl-tRNA synthetase is essential in protein synthesis as it encodes the translation factor methionyl-tRNA synthetase (MetRS) leading to the function loss of this enzyme. This may result from defective tRNAMet-aminoacylation reaction as proposed by Comisso et al.11
In conclusion, new genetic revolution tests such as whole exome and whole genome sequencing have opened doors for the identification of rare genetic causes of unexplained clinical manifestations, as in our patient.
Acknowledgment
The authors gratefully acknowledge Prince Abdullah bin Khalid Celiac Disease Research chair, Vice Deanship of Scientific Research chairs, King Saud University, Riyadh, Kingdom of Saudi Arabia, for their support.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received August 13, 2018.
- Accepted December 30, 2018.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.




{kind=link}
{kind=link}
{kind=link}