


Abstract
Objectives: To detect single nucleotide polymorphism in toll-like receptor 2 (TLR2) gene in complicated cases of measles, in order to understand the genetic basis of complex human immune responses against measles complications.
Methods: A total of 100 children consisted of 50 measles complicated cases while rest were gender matched disease-free individuals who served as controls for this study. Patient demographic data and clinical information were recorded on a separate pre-designed model form. All exonic regions of TLR2 gene of the patients and control samples were amplified through polymerase chain reaction. Various in-silico mutation verification tools like protein variation effect analyzer, MUPRO, sorting intolerant from tolerant, functional analysis through hidden Markov models, and polymorphism phenotyping v2 to study the effect of novel non-synonymous polymorphism on structure and function of TLR2 protein.
Results: Synonymous and novel non-synonymous polymorphisms were identified in measles complicated cases. Among these, rs1816702 was marked to 5 untranslated region section of TLR2 gene, while rs3804099 and rs3804100 were identified in the coding region. Novel non-synonymous polymorphisms were shown in the coding region of TLR2 gene. No significant association was established between the observed genetic polymorphisms and measles complications. However, rs3804100 increased the risk of lower respiratory tract infection.
Conclusion: The overall impact of novel non-synonymous polymorphism of TLR2 protein structure and functions was neutral and tolerated.
Measles is a highly contagious systemic viral infection caused by measles morbillivirus (MeV).1 After acquiring measles infection, suppression of immune system is the leading risk factor responsible for several measles complications like blindness, encephalitis, profuse diarrhea, otitis media, hepatitis, and myocarditis.2 Measles complications increase the risk of measles-related deaths, particularly among children under 5 years of age.3 Toll-like receptors (TLRs) are vital parts of non-specific immune responses and are members of the germ line-encoded receptors called pattern recognition receptors (PRRs), which play a significant role as primary line of defense against incursive pathogens.4 In humans, 10 TLRs (TLR 1-10) have been recognized so far. These receptors identify a wide variety of pathogen-associated molecular patterns (PAMPs).5 In the last few years, the role of TLR2 in vertebrate immunity as a first line of host defense has been emphasized. The high expression of TLR2 is restricted to some cells like macrophages, granulocytes, monocytes, and dendritic cells (DCs) of the hematopoietic system. Current research suggests signaling via TLRs that play a crucial role in the pathogenicity and immunogenicity of measles. The measles infection can trigger T-lymphocytes expression of TLR2, which consequence in cytokines production like interleukin-6 (IL-6) that augment signaling lymphocyte activated molecule (SLAM) expression that is used as a receptor by measles virus for docking process.6
It has been observed that single nucleotide polymorphism (SNP) determines a host’s susceptibility to diseases.7 For the understanding of fundamental genetic architecture of complex diseases, many strategies have been developed to comprehend the significance of polymorphism.8 The human genomic SNPs are responsible for individual variances such as phenotypic appearance, disease susceptibilities and drug metabolism.9 Genetic polymorphism phenomena may be synonymous or non-synonymous with the latter being a consequence of amino acid substitution bears the potential to alter protein structure and function.10 The location of SNP within a gene may alter the protein structure. Indeed, the SNP in the regulatory region of the gene may affect protein expression. Single nucleotide polymorphisms may impact gene splicing, binding of transcription factor and non-coding ribonucleic acid (RNA) sequence. The SNPs that are not in the proximity of the gene may be used as a genetic marker for locating a gene causing a disease.9 Toll-like receptor-2 deficient mice have been reported to be more susceptible to infection compared to wild mice. This was evident from the in-vivo study, where high titer (approximately 10 fold) of cytomegalovirus were seen in spleen, liver and lungs of TLR2 knockout mice compared to wild type.11 These SNPs may have a minor impact on the biological system, but their functional effects include variations in amino acids, stability of mRNA and transcription factor binding affinity.12 Several synonymous and nonsynonymous polymorphisms have been identified in the regulatory and coding region of TLRs and their association with infectious, inflammatory and allergic diseases.13 Severity and susceptibility of bronchiolitis, a viral infection are not only influenced by environmental factors, but host genetic heterogeneity may also play a role in determining host susceptibility. Single nucleotide polymorphism (rs7656411) in TLR2 has been associated with severe acute viral bronchiolitis in children without any co-morbidities.14 It has been observed that genetic polymorphisms in TLR2 gene affect the viral load in increased susceptibility of hepatocellular carcinoma among the patients suffering from hepatitis C virus.15 Furthermore, the presence of R753Q in TLR2 increased the risk for cytomegalovirus in post transplant liver patients.16 Non-synonymous polymorphism 1635 A/G and 1174 G/A in TLR9 were associated with rapid progression of human immunodeficiency virus (HIV) infection, while the presence of Q11L polymorphism in TLR7, increased susceptibility, accelerated progression, higher viral load and immune suppression in HIV-1 patients.17 The measles virus activates TLR2-expressing T-lymphocytes, which leads to the subsequent secretion of cytokines and pro-inflammatory markers that help in up regulation of SLAM receptors. Recognition of the measles virus through TLR2 and its specificity can be affected by the genetic polymorphism in the interacting domains of measles virus and the host cell receptor.18 Moreover, genetic polymorphism in the TLR2 gene may affect its expression, structure and impair host immune responses to a wide spectrum of pathogens.19 Many studies have indicated that the presence of SNPs in TLRs is associated with altered host susceptibility or clinical outcome to infectious diseases.20-22 The role of TLRs are very important for initial recruitment of immune cells. Understanding genetics behind enhanced predisposition of some individuals to certain measles related complications will enable us to tackle the disease through identification of high-risk individuals. Moreover, these efforts may also provide a gateway for the development of personalized medicine in future. Furthermore, the study will also provide information on how the genetic polymorphism affects the protein structure of TLR2. Therefore, keeping in mind the importance of the genetic polymorphism in the TLR2 gene with respect to its association with various diseases and inter-host susceptibility among different populations. The present study aims to identify SNP among complicated measles patients admitted to a tertiary care hospital, Peshawar, Pakistan.
Methods
The case control study was carried out from October 2019 to December 2020 at Lady Reading Hospital (LRH), Peshawar, Pakistan. We enrolled 100 children in the study, consisting of 50 complicated measles cases, and 50 age- and gender- matched disease-free individuals served as controls. All the patients were admitted to the pediatric (children) unit of LRH, which is a tertiary care hospital of this region.
Children aged >4 months but <10 years (both genders) were included. Furthermore, children with health issues like congenital heart disease, leukemia, thalassemia or other hereditary diseases were excluded from the study.
Ethical approval was obtained from the hospital’s Departmental Ethical Committee (Ref: No.218/LRH 30/9/2019). Written informed consent was attained from the guardians of all contributors.
Patients’ demographic data and clinical information such as age, feeding history, vitamin A intake, educational level of parents, and socio-economic status were recorded on a separate predesigned form. The measles vaccination status of patients was determined through their vaccination card. The sample size was calculated through OpenEpi, version 3.
A complicated measles case was considered “complicated” if a child is presenting with a non-vesicular maculopapular rash, high grade fever (that has persisted for more than 3 consecutive days) and with at least one of symptoms, namely, coryza, cough, and conjunctivitis, along with one or more complications like pneumonia, myocarditis, otitis media, sub-acute sclerosing panencephalitis or diarrhea.23 All the measles-associated complications were diagnosed by pediatric physicians on the basis of clinical scenarios and diagnostic parameters like chest x-ray, echocardiography, serum electrolytes, blood complete, and test of anti-measles immunoglobin M (IgM) from the cerebrospinal fluid (only for the encephalitis patients).
A blood sample (4 mL) was collected aseptically from both groups and approximately 2 mL was dispensed into each ethylenediamine tetraacetic acid (EDTA) and gel tube. A clear serum was obtained from the gel tube through centrifugation at high speed. Serum was separated and stored at 2-8oC for further analysis.
Measles cases were confirmed by detecting a measles-specific antibody (IgM) in the patient serum through an enzyme-linked immunosorbent assay (ELISA) method using Alegria® (a fully automated random-access analyzer, Germany). The Alegria® assay features barcoded 8-well microstrips (the Alegria® Test Strip, which hold a complete set of reagents and are designed for a single determination of one patient sample). Interpretation of the results was as follows: patient values <20 U/mL were considered negative, values between ≥20 - ≤25 U/mL were considered borderline while values >25 U/mL were considered positive.24
Genomic DNA was extracted from the blood samples (EDTA blood) in accordance with the protocol provided by Gene All ExogenTM DNA extraction kit (South Korea) and stored at -20oC for further processing.
Primer3 software was used to design the primers based on sequence information obtained from a publically available database (ensemble). Primers were designed in order to cover the whole exonic region of the TLR2 gene. All the exons were amplified with the designed primers following optimization of various conditions such as denaturation, annealing and elongation. The final polymerase chain reaction (PCR) was carried out in a 20µL reaction sample, which consisted of a 10µL master mix (2X) (Thermo Scientific, USA), 0.4µL of each forward and reverse primers, 7.2µL of PCR water and 2µL of sample DNA. After purification, all the PCR products were sent to Eurofen MWG (Operon LLC, USA) for sequencing.
Identification of SNPs was carried out using Bioedit sequence alignment editor software through clustalW multiple alignments, version 7.2.
Protein variation effect analyzer (PROVEAN), polymorphism phenotyping v2 (POLYPHEN-2), sorting intolerant from tolerant (SIFT), MUPRO and functional analysis through hidden Markov models (FATHMM) were among the mutation detection technologies employed. Protein variation effect analyzer is a new method that can predict the functional characterization of protein sequence variation. For all classes of variations, the PROVEAN score offers information on distribution of detrimental and neutral effects on variants. Variants with a score equal to or less than -2.5 (threshold value) were classified as detrimental, whereas those with a score over the threshold value were classified as neutral.25 Polyphen-2 prediction is based on the number of sequences, evolutionary structural considerations, and comparative analysis. It performs functional annotation of SNP coding maps in order to generate transcripts. Polyphen-2 quarry was qualified as either “possibly damaging” or “benign”.26 Sorting intolerant from tolerant is a multi-step technique that identifies amino acid alterations using a homology-based sequencing approach. Sorting intolerant from tolerant searched the alignment for all positions and calculated the probabilities for all amino acids in the needed positions. If a given threshold value is less than the SIFT score, SIFT predicts a modification that affects protein function. The SIFT result is either “tolerated” or “not tolerated”.27 Mupro is a support vector machine (SVM)-based approach that predicts protein stability after a single amino acid substitution by leveraging both sequence and structural information. In terms of rise or reduction, structural stability is determined. To find protein stability, a relative stability change (G) arising from a single mutation is calculated.28 A high-throughput online service called FATHMM is used to anticipate the functional consequences of non-synonymous polymorphisms and non-coding variants in the human genome. Functional analysis through hidden Markov models also aids in the analysis of large-scale genomic data sets with high throughput.29
Statistical analysis
The continuous variables were expressed as mean ± standard deviation (SD) in case of normal distribution. The Shapiro-Wilk test was used to test for normal distribution. For comparison of categorical variables, the Fisher exact test was used. The statistical testing was carried out at the 2-tailed α level of 0.05. The data was analyzed using GraphPad Prism, version 8.0.2 (GraphPad Software, Inc., USA).
Results
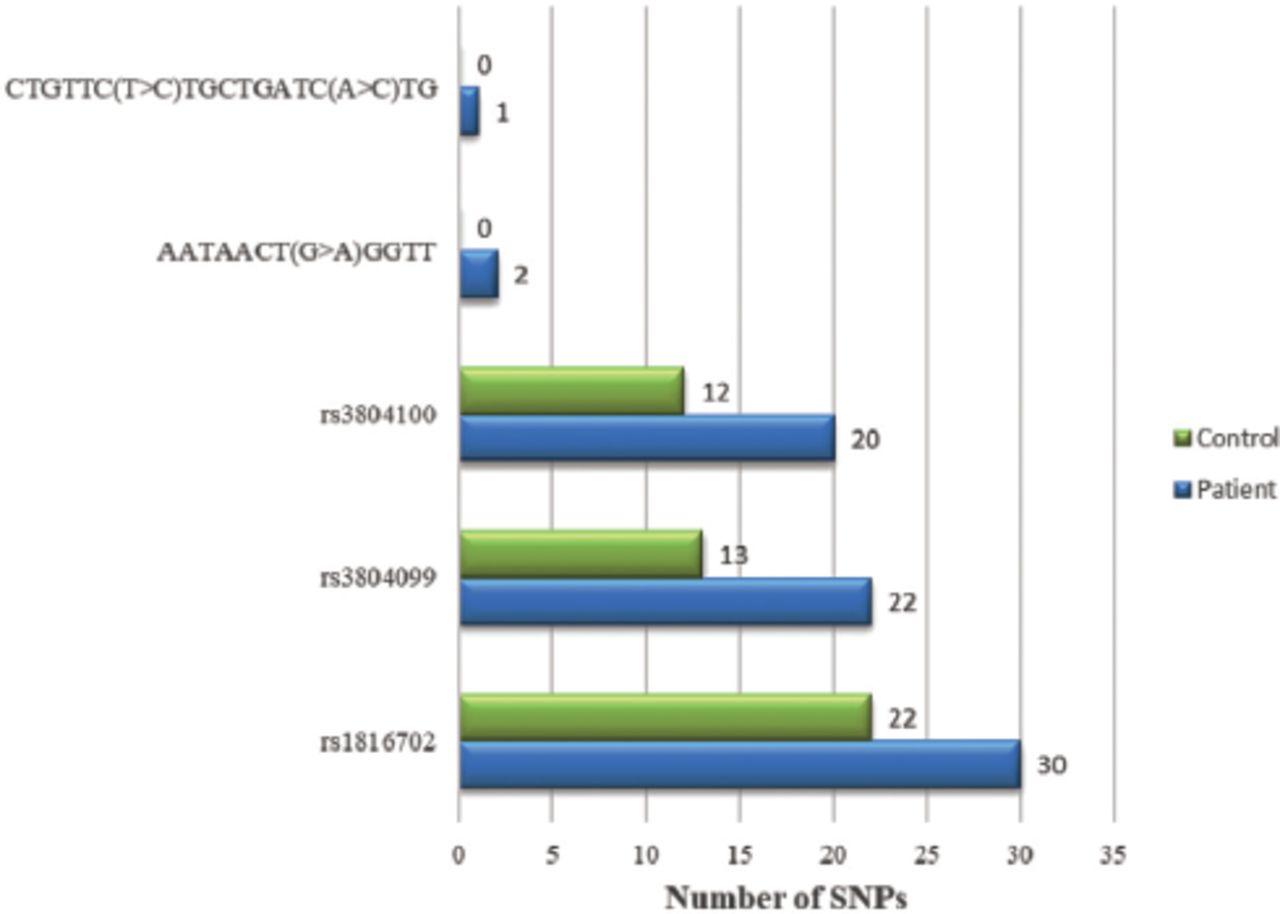
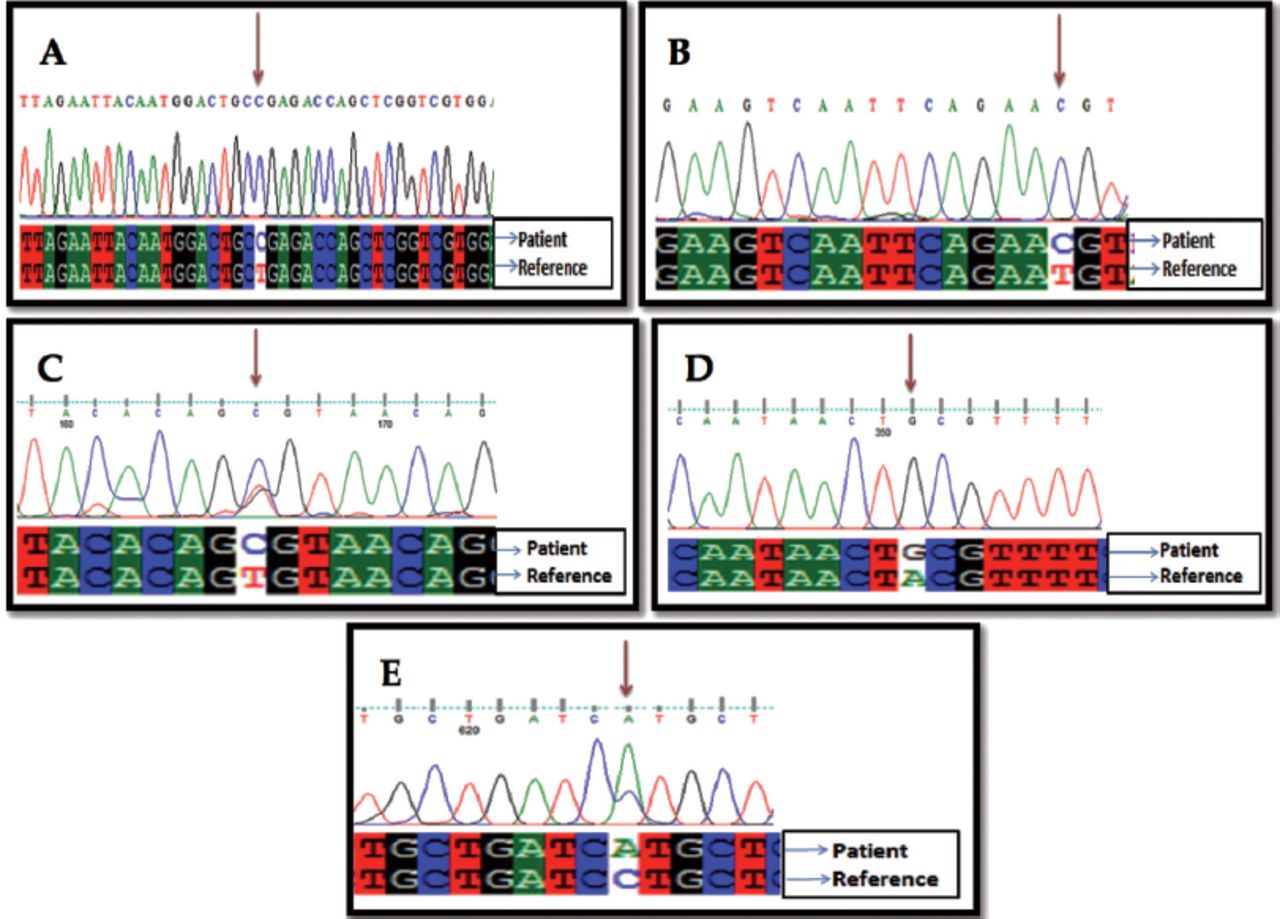
According to the demographic data of the patients illustrated in Table 1, out of 50 patients, 24 (48%) were male, with a mean age of 13.56±9.59, while 26 (52%) were female aged 14.12±8.17. Although trends are changing, breastfeeding practices are common in Peshawar, Pakistan, particularly in early childhood. It was observed that 29 (58%) of the children were breastfed, while 15 (30%) were given formula milk. A small group of babies (n=6; 12%) were fed through both methods. Measles infection was found in children (n=37; 74%) who did not receive the vaccination. Measles resurgence was observed in children (n=13; 26%) who received a single dose of the vaccine. Approximately 56% of measles-infected children received vitamin A drops during their routine vaccination schedule. The overall distribution of polymorphism in TLR2 gene among both groups is shown in Figure 1. rs1816702 was observed in 30 (60%) patients (Figure 2A). In the current study we also found 2 synonymous polymorphisms in the coding region of the TLR2 gene: rs3804099 was observed in 22 (44%) patients (Figure 2B), and rs3804100 in 20 (40%) patients (Figure 2C). In case of pneumonia, children (n=15) with rs1816702 (C>T) was greater than the genotype (T>T). However, no significant association was observed between rs1816702 (C>T) and pneumonia. The risk of lower respiratory tract infection was found to be significantly higher (p=0.004) in children carrying rs3804100 (C>T) compared to genotype (T>T; Table 2). Two novel non-synonymous polymorphisms in the coding region of the TLR2 gene have been observed. These 2 non-synonymous novel polymorphisms were found at nucleotide positions 153,704 and 444 at amino acid position 513 (Figure 2D) and at nucleotide positions 153,704 and 714 for amino acid position 603 (Figure 2E & Table 3). According to the PROVEAN score for T513A (-0.517) and L603 (-962) the overall effect was neutral. Similarly, predictions obtained from other verification tools such as SIFT, FATHMM and POLYPHEN-2 are shown in Table 4. The MUPRO results suggest a relative change in the energy level (T513A = -0.906, L603M = -1.458), which decreases the stability of the protein structure. However, the overall impact of novel polymorphism on the protein structure and functions of TLR2 was non deleterious and not damaging.
- Demographic data and related information of the patient (n=50) and control (n=50) subjects.

- Distribution of single nucleotide polymorphisms (SNPs) in patient and control group.

- Chromatogram of rs1816702 (A), rs3804099 (B), rs38040100 (C), novel T513A (D), and novel L603M (E).
- Association of measles complications with SNPs in the TLR2 gene.
- Synonymous and non-synonymous polymorphisms in the TLR2 gene.
- Effect of non-synonymous polymorphisms on structure, function and stability of TLR2 protein.
Discussion
Measles is a highly communicable disease that increases the risk of morbidity and mortality in children, particularly among under developed nations. A measles infection can lead to serious consequences that can be fatal.30 In the present study measles and its complications were observed in early childhood patients of this region. Furthermore, the majority of the pediatric patients (64%) with measles complications were found to be unvaccinated. Immunosuppression caused by measles allows for subsequent bacterial infection, which increases morbidity and death in young children.31 The presence of a genetic polymorphism in the immune response gene involved in TLR2-dependent signaling could influence the expression of various cytokines level, alteration in the level of cytokines leads to immunosuppression particularly in measles infection.32 Toll-like receptor-2 is an important member of the pathogen recognition receptors and its presence in human cells and tissues exerts a pivotal role in the first line of host defense.20 A number of viral proteins derived from cytomegalovirus, herpes simplex virus, hepatitis C virus, and measles virus are sensed by TLR2.33 However, genetic variations in the TLR2 gene determine the risk of severe infections in humans, mice and domestic animals.34 In the present study 2 major secondary infections, bronchial pneumonia and lower respiratory tract infection observed among measles-infected children were common measles complications, which increase the risk of mortality in early childhood. The number of children suffering from bronchial pneumonia and lower respiratory tract infection having rs1816702 (C>T) was higher than genotype (T>T). However, no significant association was established between rs1816702 and measles complications. In a previous study, the presence of rs1816702 in Danish patients was found to increase the risk of Crohn’s disease.35 The SNPs present in the regulatory region may affect the expression function of the gene. They also affect gene splicing, transcription factor binding, and the sequencing of non-coding RNA.9 According to previous data, synonymous polymorphisms have a minimal effect on the biological system and are implicated in diseases. Perturbation of mRNA splicing, the stability of the mRNA through cis factor and mRNA structure modulation are the mechanisms by which synonymous polymorphisms exert their impact on gene function.36-38 In the present study we also found synonymous polymorphism in the coding region of the TLR2 gene. In the present study we observed rs3804099 (genotype C>T) in children suffering from bronchial pneumonia, a serious fatal measles complication. However, no significant association was found between rs3804099 and pneumonia. Although in a previous study, the presence of rs3804099 was reported to play an important role in decreasing the risk of hepatocellular carcinoma.39 This disagreement between the current and previous study might be attributed to the small sample size. In the present study, we have identified rs3804100 in TLR2 and found it to be associated with measles complication. It has been observed that rs3804100 (C>T) increased the risk of lower respiratory tract infection (p=0.004) compared to genotype (T>T). In the current study, we observed 2 novel non-synonymous polymorphisms in the coding region of the TLR2 gene. These 2 non-synonymous polymorphisms substituted amino acids at positions 513 and 603 in the coding region of TLR2 protein. Both novel polymorphisms were submitted to online in-silico mutation verification tools such as PROVEAN, MUPRO, SIFT, FATHMM and POLYPHEN-2. These tools were used to evaluate the influence of non-synonymous polymorphism on protein structure and function. Polyphen-2 is a computer program that predicts how changes in protein will affect the protein’s structure and function. These kinds of automatic predictions are important for interpreting large data sets of rare genetic variants. In the current study, we also applied these automatic tools for these novel variants. According to the predictions obtained from these verification tools, the overall impact of novel non-synonymous polymorphism on TLR2 protein structure and functions was neutral and not damaging. However, the implication of synonymous and non-synonymous polymorphism in the human genome may determine a host’s susceptibility to various diseases. Furthermore, the impact of any single allele on disease outcome is often modest.
Study limitations
This study has a small sample size. The findings of this study further necessitate establishment of associations between genetic polymorphisms and measles complications. Furthermore, it is recommended that more convincing association studies of polymorphisms with diseases and complex inheritance patterns should include large sample sizes, statistical adjustments for multiple comparisons, replication of findings with independent cohorts, multiple study designs, adjusting the data for population admixture, as well as precise genetic and cellular investigations, will all be crucial in this regard.
In conclusion, rs1816702 was found in the regulatory region while rs3804099 and rs3804100 were found in the coding region of the TLR2 gene. The overall effect of novel non-synonymous polymorphism on protein structure and functions of TLR2 was neutral and tolerated. The major outcome of this study is the identification of synonymous and non-synonymous polymorphism in TLR2 gene among complicated measles cases. The presence of synonymous polymorphism in TLR2 may alter host susceptibility and increased the risk for secondary infections, while non-synonymous polymorphisms have no lethal effect on protein structure of TLR2.
Acknowledgment
The authors gratefully acknowledge Mr. Richard Thift of English Editing Netherland, registered at the Amsterdam Chamber of Commerce (KvK) as ‘Amsterdam English Lessons’ (KvK number: 56231377. VAT number: NL002526762B89) for English language editing.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received June 26, 2021.
- Accepted October 5, 2021.
- Copyright: © Saudi Medical Journal
This is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
References
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.