


Abstract
Woodhouse-Sakati syndrome (WSS) is a rare genetic condition of autosomal recessive inheritance pattern. The disease is characterized by a group of disorders, including diabetes mellitus, alopecia, hypogonadism, intellectual disability, and progressive extrapyramidal signs. This syndrome is related to an inherited neurodegenerative disorder’s heterogeneous group characterized by the accumulation of iron in the brain, caused by a mutation in the DCAF17 gene. This report discusses the case of 3 Saudi sisters having WWS. The 3 sisters aged 18, 22, and 25 years took birth to consanguineous parents (first-degree cousins). The sisters initially had normal developmental growth with deprived scholastic performance because of the intellectual difficulties. At puberty, the secondary sexual characteristics were not developed in the patients, and they faced primary amenorrhea. They were found to have features typical of WSS, but they also had gynecological anomalies, which are considered unusual findings in WSS patients.
Woodhouse Sakati syndrome (WSS) is an uncommon disorder of autosomal recessive neuroendocrine, further categorized by the extrapyramidal features, sensory neural deafness, mental retardation, diabetes mellitus (DM), hypogonadism, and alopecia.1 Most of the reported families described in the medical literature are from Middle Eastern countries. Until now, almost 32 families (involving 23 with molecularly confirmed diagnoses) are reported, including 76 total individuals who have been affected.1 None of those cases reported had gynecological anomalies. Congenital anomalies among the genital tract of the female are uncommon but significant diagnoses that may present during adolescence or childhood. Fewer hormones are produced in patients with WWS, affecting their sexual development (hypogonadism) that usually becomes visible in adolescence and most probably have a rule in congenital malformations of the female genital tract.
The individuals affected by WSS possess distinctive dysmorphic features categorized by hypertelorism, prominent nasal bridge, and long triangular face. It also consists of predominant neuroendocrine manifestations and progressive nature, which become frequent during early adulthood and adolescence. Fundamentally, every individual will possess endocrine outcome as the early manifestation of WSS, which is progressive childhood-onset hair thinning. Every patient suffers from mixed-nature hypogonadism. Above-average affected individuals will possess variable neurologic manifestations involving bilateral post-lingual sensorineural hearing loss, mild intellectual disability, and progressive extrapyramidal movements (namely, dystonic spasms with dysphagia, dysarthria, and dystonic posturing). Fewer recognized manifestations involve electrocardiographic abnormalities, keratoconus, and seizures.2 The aim of this study was to discuss the cases of 3 Saudi sisters having WWS.
Case Report
A genetic analysis was carried out at King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia. Deoxyribonucleic acid (DNA) was extracted from peripheral leukocytes according to standard procedures. All coding exons of the DCAF17 gene were amplified using polymerase chain reaction (PCR). Sequencing analysis of DNA revealed the homozygous mutation c.436delC in exon 4 of the DCAF17 gene, confirming WSS diagnosis.
This report discusses the cases of 3 Saudi sisters with WWS aged 18, 22, and 24 years. Table 1 summarizes the main clinical features of the patients.
- Comparison of our cases’ clinical features with clinically diagnosed cases reported in literature.
Patient one
The first patient (Figure 1) was 18 years old presented to the endocrine clinic because of delayed puberty.

- Timeline with a brief description and follow up of the 3 cases.
Clinical finding
She had normal developmental growth and antenatal and postnatal stages but poor scholastic performance because of perceived intellectual difficulties. She dropped out soon after her school’s third year. At the puberty age, her secondary sexual characteristics were not developed. She had mild extrapyramidal symptoms and primary amenorrhea. At this time, a complete hormonal profile was suggested, which made sure that she had gonadal dysfunction, DM, and hypothyroidism.
Physical examination showed no secondary sexual characteristics, a tall stature, triangular face, and frontotemporal alopecia. The patient had breast hypoplasia and had no pubic or axillary hair. Her neurological examination was unremarkable, and her gynecological examination revealed abnormal female sex organs. She had normal labia majora, with slight swelling, and a normal clitoris. She had no labia minora, and there was one wide opening with the urethra inside and no vaginal orifice.
Diagnostic assessment
Regarding the laboratory investigations, the hormonal profile of the patient showed high thyroid-stimulating hormone (TSH) (8.27 mIU/L, normal 0.4-4 mIU/L), normal growth hormone (0.337 ng/mL, normal <10 ng/mL), low free thyroxine (FT4) (10.26 mIU/L, normal 9-25 mIU/L), high follicle-stimulating hormone (18 mIU/L, normal range 3.03 - 8.08 mIU/L), high luteinizing hormone (11.9 mIU/L, normal range 1.8-11.78 mIU/L), high glycated hemoglobin (HbA1c) (7.1%, normal 4-5.6%), low cortisol (107 nmol, normal 118-618 nmol), normal adrenocorticotropic hormone (ACTH) (12 pcg/mL, normal 9-52 pcg/ml), normal prolactin (106.21 µIU/ml, normal range 102-469 µIU/ml), low estradiol (5 pg/ml, normal 7.63-42.6 pg/ml), low dehydroepiandrosterone (DHEA) (68.69 mcg/dL, normal range 148-407 mcg/dL), low testosterone (0.087 nmol/l, normal 0.29-1.67 nmol/l). These data from the laboratory are suggestive of hypergonadotropic hypogonadism, hypothyroidism and DM.
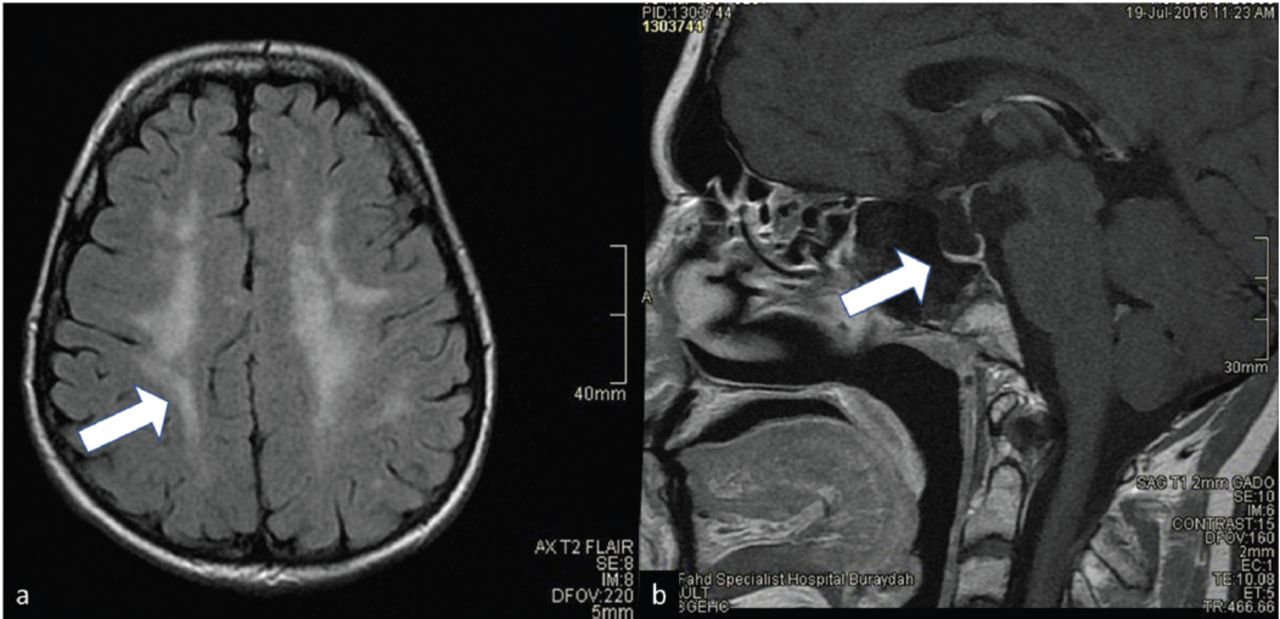
Magnetic resonance imaging (MRI) of the pituitary (Figure 2) showed that the size of the sella was normal, measuring 12 mm in the maximum anteroposterior dimension. The size of the pituitary gland was small for the patient’s age and gender, and it appeared to be flattened against the sellar floor. It measured 1 mm in the maximum craniocaudal dimension. The bright focus of the posterior pituitary lobe was reduced. The pituitary stalk was reduced in volume. The suprasellar cistern was preserved with no evidence of compression of the optic chiasm. The cavernous sinuses were normal. The flow voids of the cavernous segments of the internal carotid arteries were preserved. There were bilateral and asymmetrical diffuse and confluent white matter diseases in the brain with T2 and FLAIR hyperintensity without corresponding diffusion restriction or abnormal enhancement. The subcortical U fibers were preserved. Infratentorial involvement was also evident at the level of the pons and brachium pontis. The septum pellucidum was preserved, and the corpus callosum was normal. The chiasm and optic nerve volume were preserved. There was no marked reduction in the T2 signal in the substantia nigra and basal ganglia to suggest iron deposition. There was no abnormal parenchymal or meningeal enhancement. There were minor inflammatory changes in the ethmoidal air cells. All of these are features of pituitary hypoplasia and white matter disease in a patient with WSS.2 The pelvic MRI displayed no ovaries or uterus; the vagina was short and opened at the base of the urinary bladder, which had slightly thick walls.

- Magnetic resonance imaging for the brain of case number 1. A) T2 flair showing white matter disease. B) T1 image showing pituitary hypoplasia.
Patient 2
Twenty-two years old, and at the age of 20, she was diagnosed with DM with bilateral visual abnormalities because of the keratoconus.
Clinical finding
She was brought for examination by her mother because of delayed puberty and primary amenorrhea. Physical examination showed alopecia totalis and had breast hypoplasia. The patient had no pubic or axillary hair and no secondary sexual characteristics. Her neurological examination was unremarkable, and the gynecological examination revealed abnormal female sex organs. She had normal labia majora with slight swelling, a normal clitoris, no labia minora, and one wide opening with the urethra inside with a tiny vaginal orifice.
Diagnostic assessment
The relevant laboratory findings were as follows: HbA1c level of 7.3%, a high TSH level of 4.33mIU/L, a low FT4 level of 8.41, a high LH level of 14.7133mIU/L, a high FSH level of 31.8133mIU/L, a low level of testosterone as 0.224 nmoL/L and an average prolactin level of 25033 mIU/L.
Pelvic MRI showed no ovaries or uterus; the vagina was short and opened at the base of the urinary bladder, which had slightly thick walls. The MRI of the pituitary showed that the size of the sella was normal, measuring 9 mm in the maximum anteroposterior dimension. The pituitary gland was small for the patient’s age and gender, and it appeared to be flattened against the sellar floor, measuring 3 mm in the maximum craniocaudal dimension. The bright focus of the posterior pituitary lobe was reduced and normal. The pituitary stalk was reduced in volume. The suprasellar cistern was preserved with no evidence of compression of the optic chiasm. The cavernous sinuses were normal. The flow voids of the cavernous segments of the internal carotid arteries were preserved. In the brain, there were only 2 punctate foci of T2 and FLAIR hyperintensity in the periventricular and subcortical white matter regions of the frontal lobe, without corresponding diffusion restriction or abnormal enhancement. The subcortical U fibers were preserved. No infratentorial involvement was observed. The septum pellucidum was preserved, and the corpus callosum was normal. The chiasm and optic nerve volume were preserved. No marked reduction in the T2 signal in the basal ganglia and substantia nigra was observed to suggest iron deposition. There was no abnormal parenchymal or meningeal enhancement. There were minor inflammatory changes in the ethmoidal air cells. All of these are features of pituitary hypoplasia and white matter disease in a patient with WSS.2
Patient 3
Twenty-four years old and had been diagnosed with DM at the age of 17. The patient had also been diagnosed with hypothyroidism at the age of 19 and was receiving treatment with metformin.
Clinical finding
The patient was brought for examination because of delayed puberty and primary amenorrhea. Physical examination showed a long, triangular face, alopecia totalis, a high arch to her palate, and low intellectual ability. The absence of secondary sexual characteristics was also determined. She had breast hypoplasia, and no pubic or axillary hair was present. Her neurological examination was unremarkable. Her gynecological examination revealed abnormal female sex organs. She had normal labia majora with slight swelling, a normal clitoris, no labia minora, and one wide opening with the urethra inside without a vaginal orifice.
Diagnostic assessment
The relevant laboratory findings were as follows: HbA1c level of 12.8, normal TSH level of 3.133mIU/L, low FT4 level of 9.58, high LH level of 11.933mIU/L, high FSH level of 2833mIU/L, normal prolactin level of 315mIU/L, the low testosterone level of 0.27nmol/l, and low estradiol level of 25pg/ml.
Pelvic MRI showed no uterus or ovaries; the vagina was short and opened at the base of the urinary bladder, which had slightly thick walls. The results of the MRI of the pituitary were the same as those for the second sister. The results indicated pituitary hypoplasia with white matter changes suggestive of WSS.
Therapeutic intervention
The 3 cases were managed supportively, aiming to control DM, and hypothyroidism. The patients refused hormonal replacement therapy to promote bone health. Genetic counseling was carried out.
Follow-up and outcomes
All patients were followed regularly at the clinic to monitor for endocrine abnormalities. Therapeutic intervention for the concurrent anomalies needs a multidisciplinary team and more research because of the lack of evidence.
Discussion
Woodhouse Sakati syndrome is an uncommon or rare disorder; which possesses unknown prevalence. Few dozen families that are affected, from the Middle East, have been explained in the medical literature. Initially determined in 1983, this is an uncommon disorder of autosomal recessive gene inheritance that further leads to endocrinal symptom spectrum. Most of the reported families are from the Middle Eastern countries described in the medical literature. Until now, almost 32 families (involving 23 with molecularly confirmed diagnoses) are reported, including 76 total individuals who have been affected.1 The responsible gene, DCAF17, is present on chromosome 2q31.1.5
Woodhouse Sakati syndrome is categorized by DM, intellectual disability, hearing loss, hypothyroidism, hypogonadism, and alopecia. Electrocardiogram anomalies have also been described.5 Several WWS manifestations, such as neurological and endocrinological disorder, and alopecia have been reported. Most manifestations appear later in life, as was the case in these 3 cases, who presented with alopecia, DM, hypothyroidism, extrapyramidal symptoms, and intellectual disability. However, the gynecological anomalies observed in these sisters are novel findings with unknown mechanism.
The hypogonadism nature in these patients has been problematic to explain because hypogonadotropic and hypergonadotropic hypogonadism, both have been defined.5 In approximately 30% of the total affected individuals, the profile of hormones does not fit either group. Hypergonadotropic hypogonadism was identified in our patients.
Women can have primary amenorrhea, which can cause concern on the part of the patient and the patient’s family.6 Detailed endocrine investigations, which were available for 52 females explained in the literature, showed no estradiol or extremely reduced levels and high levels of LH, and FSH, which are stable with hypergonadotropic hypogonadism, as was observed in our patients. There appears to be decreased hypothalamic-pituitary responsiveness, as the FSH, and LH levels are not as high as expected given degree of ovarian failure.6
One of the affected sisters had bilateral keratoconus, which has been reported as a symptom of WSS.7
The brain MRI findings met the described radiological criteria, of which are white matter lesions and pituitary hypoplasia.8 The described outcomes can moreover distinguish WWS from different types of other neurodegenerative diseases with subtypes of brain iron accumulation.
The study of genetics directed by Alazami et al,2 on 15 families assisted in the mutation identification in the DCAF17 gene, which causes WSS. The mutations found in our 3 cases were pathogenic.
In conclusion, WSS is widely prevalent with different manifestations reported. However, based on the cases presented in this study, gynecological anomalies might be considered pathological findings in patients with WSS. Further studies are needed to investigate treatment options.
Acknowledgment
The authors gratefully acknowledge Dr. Ali Alamer, Neuroradiologist consultant, Radiology Department, Qassim University, Qassim, Saudi Arabia, for his effort in reviewing MRI images for correctness and clarity. We also would like to thank Manuscript Edit (https://www.aje.com/) for English language editing.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received July 26, 2021.
- Accepted October 8, 2021.
- Copyright: © Saudi Medical Journal
This is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.