


Abstract
We report a case involving a 73-year-old Saudi man diagnosed with dermatomyositis who subsequently developed Kaposi’s sarcoma one month later. He had difficulty in rising from a chair and increased leg weakness while climbing stairs or walking. He was unable to comb his hair and had greater dysphagia with liquids than with solid foods. Laboratory tests showed elevated liver enzyme and creatine kinase levels. Right quadriceps muscle biopsy indicated inflammatory myopathy, which was consistent with adult dermatomyositis. We administered 1-g/day methylprednisolone intravenously for 3 days, followed by 60-mg oral prednisolone daily, which led to significant improvements in his muscle strength and dysphagia symptoms. The creatine phosphokinase level normalized. Computed tomography and endoscopic examinations did not reveal any evidence of an underlying malignancy. The patient was discharged and underwent close monitoring at a rheumatology clinic. We subsequently added 50-mg oral azathioprine daily to the treatment regimen. At one month after emergency room presentation, multiple new dusky violaceous macules and plaques appeared on his chest, face, upper back, neck, and upper limbs. A biopsy of one of these lesions showed findings consistent with
Kaposi’s sarcoma. He was referred to an oncology center for chemotherapy, following which his skin lesions significantly regressed. Dermatomyositis may be considered a paraneoplastic syndrome of Kaposi’s sarcoma.
Kaposi’s sarcoma (KS) is one of the vascular tumors usually associated with human herpes virus 8 (HHV-8) infection, and is most commonly found among immunocompromised patients, mostly those with human immunodeficiency virus (HIV) and those who have undergone organ transplantation. Dermatomyositis (DM), although associated with other malignances, has rarely been described in association with KS in the previous literature. While these KS cases describe cutaneous involvement, disseminated Kaposi’s sarcoma has not frequently been reported. We report the case of a 73-year-old man diagnosed with adult DM who subsequently developed disseminated cutaneous KS at one month after the adult DM diagnosis, thus raising the possibility of the major role of immunosuppression in promoting the progression of KS rather than the consequence of his immunosuppressive medications, such as a steroid or low-dose azathioprine. We reviewed the literature of the previous clinical cases of DM with similar association and provided the possible underlying mechanism of the development of KS. Finally, we discussed the appropriate management in such conditions, as there is no firm established guideline in the literature for similar cases. Ethics committee approval was taken, and informed written consent was obtained from the patient to have his data published along with the accompanying images.
Case Report
Patient’s information
Our patient was a 73-year-old man without a significant medical history, who presented to our emergency department with complaints of difficulty in standing up from a sitting position and difficulty in raising his arms when brushing his teeth or combing his hair for a duration of 7 days. These symptoms were also associated with greater difficulties in swallowing fluids than in swallowing solid foods. His son noticed skin rashes on his hands, face, upper back, upper chest, and thighs. There was no history of blood transfusion, drug abuse, or high-risk sexual behavior.
Clinical findings
His vital signs were stable, and systemic examination showed unremarkable findings, apart from his skin examination that revealed significant heliotrope rash, Gottron’s papules, Gottron’s sign, V-sign, and shawl sign, which are typical skin findings for DM. His motor neurological examination revealed a significant proximal myopathy with a muscle power grade of one on a 5-grade scale.
Diagnostic assessment
Results of the laboratory tests showed elevated C-reactive protein, creatinine kinase (CK), and muscle enzyme levels as well as positivity for antinuclear antibody (1:320 homogeneous) and negativity for myositis antibodies, anti-Mi-2, and anti-Jo-1 (Table 1). He was admitted to the medical ward, and normal saline was administered intravenously. Magnetic resonance imaging of the bilateral thighs showed findings consistent with inflammatory myopathy. A right quadriceps muscle biopsy revealed inflammatory myopathy consistent with DM. An electromyogram of his right shoulder showed results suggestive of a myositis pattern. The tumor marker tests demonstrated negative results (Table 1).
- Laboratory data at diagnosis of dermatomyositis and at 2 months post-treatment.
Therapeutic intervention
He was started on intravenous (IV) methylprednisolone at a dose of 1 g per day for 3 days as a pulse therapy, especially that he has esophagus involvement, followed by oral prednisolone at a dose of 60 mg daily. This treatment led to a significant improvement in his muscle strength. His serum creatine phosphokinase levels returned to normal after one week of treatment. His condition was complicated with aspiration pneumonia secondary to his significant dysphagia, which required intravenous antibiotic treatment (IV piperacillin/tazobactam 4.5 g every 8 hours for 7 days); he showed great recovery after receiving this treatment. He underwent chest, abdominal, and pelvic computed tomography (CT), as well as upper and lower endoscopy, which did not reveal any evidence of an underlying malignancy. However, high-resolution CT of his chest revealed mild traction bronchiectasis with central distribution on the bilateral lower lobes and a fibrotic non-specific interstitial pneumonia pattern, which was likely a sequela of a collagen vascular disease. The patient was discharged from the hospital after 10 days and underwent close monitoring at a rheumatology clinic. Pulmonary function test was carried out during an outpatient visit, and all parameters were within normal range. Low-dose oral azathioprine at 50 mg once daily was subsequently added to the treatment regimen after 2 weeks from the patient’s presentation to our emergency department as a disease modifying anti-rheumatic drugs. At one month after the initial presentation, multiple new dusky violaceous macules and plaques appeared on his face, chest, upper back, neck, and upper limbs (Figure 1). A biopsy of one of these lesions showed findings consistent with KS. The HIV antibody test (enzyme-linked immunosorbent assay [ELISA]) showed negative results. We discontinued his treatment with azathioprine, and oral prednisolone was tapered down quickly to the lowest tolerable dose of 5 mg/day; no recurrence of his myopathy occurred after these changes in the treatment regimen. We immediately referred him to the nearest oncology center as he continued to develop more new skin lesions despite these changes in his treatment regimen. He received 16 sessions of chemotherapy (Doxorubicine 20 mg /m2 IV every 2 weeks), as his primary oncologist considered him as having disseminated cutaneous KS associated with DM.
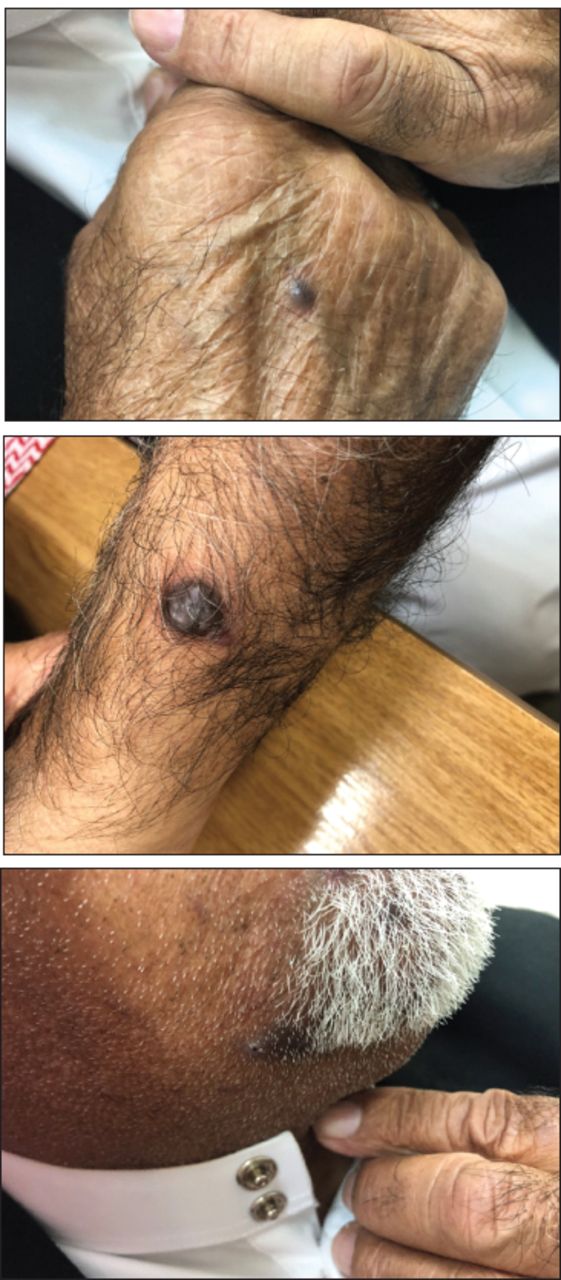
- Appearance of multiple violaceous oval and round macules and plaques on the patient’s face and upper limbs.
Follow-up and outcomes
At 6 months after the initial event, the patient was noted to have marked regression of his cutaneous lesions. One year later, given that his conditions remained stable while on treatment with oral prednisolone at a dose of 5 mg once daily, and he continued to have normal CK levels with no evidence of myopathy or any new skin lesions related to his previous KS, we elected to taper his prednisolone dose gradually by 1 mg every 4-6 weeks until discontinuation. A summary of the patient’s history and timeline of the outcomes are given in Table 2.
- Timeline of the outcomes of our patient from the initial visit to the treatment and follow-up visits.
Discussion
Kaposi’s sarcoma is the most common malignancy found in HIV patients, which involves the skin, mucous membranes, visceral organs, and lymph nodes. The etiology of KS is multifactorial, and the combination of a hereditary predisposition and immunological factors resulted in a major increase in the risk of developing this disease.1 Most of the existing literature is based on the authors’ experience with patients having HIV. Multiple cases have been reported in the literature, which were mostly associated with renal transplantation; although not quite common, KS cases associated with connective tissue diseases, such as rheumatoid arthritis, systemic lupus erythematosus, polymyositis, and DM, were also reported.2,3 Most of these KS cases that were associated with immunosuppressive medications used in the treatment of the connective tissue diseases are usually limited to cutaneous involvement; however, there are several cases of diffuse visceral involvement occurring in patients who had undergone renal transplantation and received multiple immunosuppressive medications.4,5 In our patient, KS may be a paraneoplastic sign, rather than the consequence of immunosuppressive therapy, because, among the previous rheumatologic patients treated with steroid, the time interval between the initiation of steroid therapy to the development of KS ranged from 3 months to >36 months, with the average duration being 13.7 months. Moreover, our patient developed KS earlier than the reported time period. He developed KS at one month after the initiation of oral prednisolone and at 2 weeks after starting a low dose of Imuran at a low dose of 50 mg once daily, which may indicate that the KS development in our case was most likely a paraneoplastic syndrome, rather than the consequence of immunosuppressive therapy.
Generally, corticosteroid withdrawal or reduction results in clinical regression of KS. However, when the tumor does not regress and disseminated skin lesions or visceral involvement is present, antitumor therapy, including chemotherapy and/or radiation therapy may be required; this is similar to our case, with our patient being considered as having KS with disseminated skin involvement. Moreover, another interesting point that should be considered is that if the reduction of oral steroid dose and cessation of azathioprine did not result in tumor regression, but rather led to an increase in tumor growth, it would further strengthen the theory that the KS development was a paraneoplastic syndrome rather than the result of the immunosuppressive therapy. Fortunately, our patient responded very well to the chemotherapy treatment with no relapses of his DM even after the discontinuation of the oral steroid treatment.
In conclusion, although cases of DM with KS rarely occur, DM may be considered a paraneoplastic syndrome of KS, especially if KS lesions appear before DM, <3 months after the diagnosis of DM, or before starting the immunosuppressive therapy, as this treatment may play an essential role in promoting the progression of the KS. Thus, clinicians should be cautious when starting immunosuppressive therapies in patients with DM, especially in elderly patients, as it may promote the progression of KS and other related malignancies.
Acknowledgment
We would like to thank Editage for the English language editing of our paper and the Saudi Society for Rheumatology for providing financial support for English language editing of our paper.
Footnotes
Disclosure. Authors have no conflict of interests, and the work was not supported or funded by any drug company.
- Received September 23, 2020.
- Accepted February 10, 2021.
- Copyright: © Saudi Medical Journal
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial License (CC BY-NC), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.




{kind=link}