


Abstract
Objectives: To compare Ras-related associated with diabetes (RRAD) across different species and to identify specific biomarkers for cancer therapy.
Methods: The study involves comparing the coding sequences, genes, messenger ribonucleic acid (RNA), non-coding RNA, open reading frame, short- and long-sequence repeats, and transcription factors of RRAD genes from 82 species. Various tools and software are employed for these comparisons, and evolutionary analysis was carried out to understand the gene’s evolutionary history. The data are classified based on forward and reverse sequences.
Results: Our analysis indicates that ACTG1 may function as a downstream effector of RRAD, offering potential avenues for diabetes and cancer treatments. By collecting RRAD sequences from 82 species and carrying out comparative genomics, this study provides diverse strategies for developing biomarker-based therapeutics. Furthermore, it suggests using RRAD in other organisms as a model for studying the knockdown effects of specific sequence sets. The study presents RRAD sequences from 82 organisms across different families, contributing to a diverse knowledge base for identifying drug-designing biomarkers.
Conclusion: This research offers insights into the potential of RRAD as a therapeutic target in various organisms and highlights the importance of biomarker identification in drug development.
In disease, hepatocellular carcinoma (HCC) is one of the deadliest malignancies worldwide. Ras-related associated with diabetes (RRAD) is one of the Ras-related guanosine triphosphatases (GTPase) that have been embroiled in metabolic disease and several types of malignant growth. This negative regulation of NF-kappa B signaling is associated with poor prognosis and tumor progression. The RRAD is under expressed in tumors, where it functions as a tumor suppressor. Conversely, its overexpression has been observed in the skeletal muscle of individuals with type II diabetes. The RRAD interacts with various downstream effectors, impacting cellular functions such as tumor cell motility, apoptosis, and metabolism.1-3 Rad is a Ras-related small GTPase that appears to repress malignant growth cell division, and its expression is often not expressed or downregulated in lung disease cells.1,2,4-6 The RRAD adversely directs NF-κB motioning to hinder the glucose transporter 1 (GLUT1) translocation and the Warburg impact in lung cancer growth cells. Hindering NF-κB flagging generally cancels the inhibitory effects of RRAD on the translocation of GLUT1 to the plasma layer and the Warburg impact. Studies have uncovered a novel component by which RRAD controls the Warburg impact in lung disease cells.7-9 Ras-related diabetes articulation is upregulated in all of the control modes and increments with age in human skin and fat tissues. Tumor protein 53 (p53) and NF-κB tie to RRAD genomic locales and adjust RRAD translation. The p53 genes suppresses RRAD genes’ activity, leading to insulin resistance development.10,11 Studies have proposed RRAD as a biomarker and a novel negative regulator of cell senescence.12 Rad can control the NFκB pathway, and the upregulation of Rad in cells brings down both the basal and TNFα-invigorated transcriptional movement of NFκB. Ras and related G proteins are relied upon for more broad applications to different compounds that catalyze phosphoryl (-PO(3)2-), including kinases and phosphatases.13-16 To comprehend the molecular mechanisms of hidden bortezomib obstruction, we should recognize the potential objective genes differentially expressed in bortezomib-safe leukemia cells versus organ controls.17-20 In this study, we extricate the short sequence repeats (SSRs) to comprehend this issue and focus on the medication component with new methodologies. Previous investigations proposed that the outflow of RRAD is downregulated in HCC tissues in contrast to the adjoining non-tumorous liver tissues in messenger ribonucleic acid (mRNA) and protein levels. Lower RRAD regulation has, thus, become an independent prognostic marker for the endurance of HCC patients.6,21 Moreover, RRAD restrains hepatoma cell oxygen-consuming glycolysis by contrarily controlling the activity of GLUT1 and hexokinase II. What is more, RRAD restraint significantly expands hepatoma cell intrusion and metastasis. The abnormal methylation of RRAD might be engaged with the pathogenesis of a subset of esophageal squamous cell carcinoma. The mRNA of RRAD is communicated mostly in the skeletal and cardiovascular muscle of people with type II diabetes when contrasted with regular health conditions.5,22,23
The RRAD gene, associated with Ras-related GTPase activity, has implications in HCC. Studies suggest that dysregulation of RRAD can impact glucose metabolism, specifically glycolysis, which plays a crucial role in the rapid proliferation of cancer cells, including those in HCC. Our study delved into a comprehensive analysis of RRAD across various species.3,11,24,25
The aim of this study is to extensively analyze the RRAD gene of different organisms for the predictive classification of sequence length, coding sequence (CDS) analysis, mRNA analysis, non-coding RNAs (ncRNA), the open reading frame (ORF), repeat region, transcriptional factors (TFs), phylogenetic analysis, and comparative studies to understand whether the genes show similarities and evolutionary deviations and to identify the significant components of the genes that encode RRAD.
Methods
We first searched the PubMed and Scopus databases comprehensively for research summarizing all RRAD for comparative evolutionary research. The methodology for this study is illustrated in the flowchart diagram shown in Figure 1A. A variety of molecular techniques were employed to carry out the necessary molecular and genetic analyses.
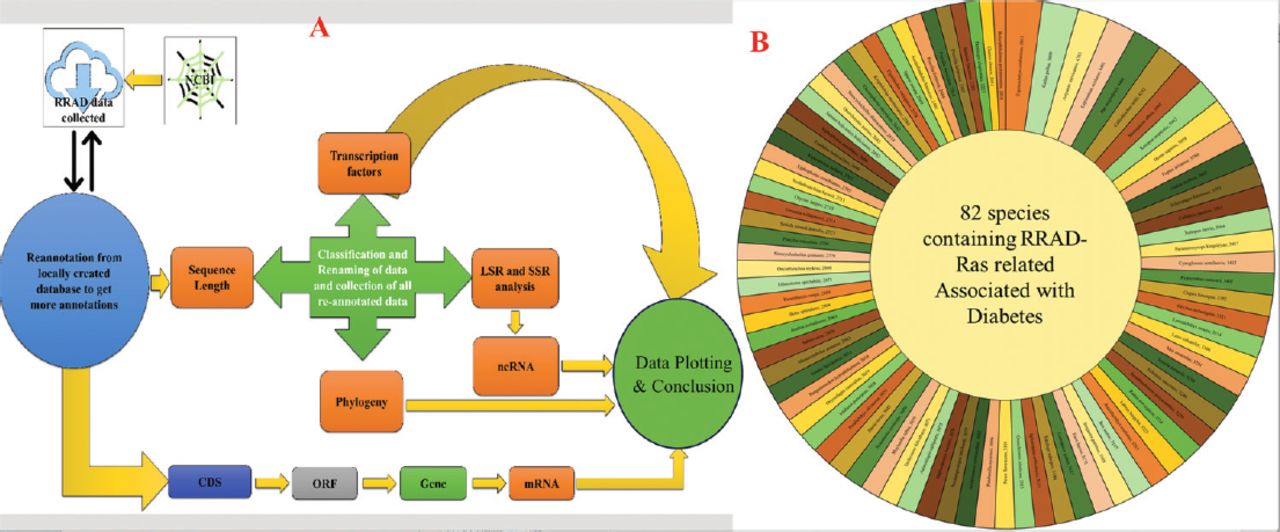
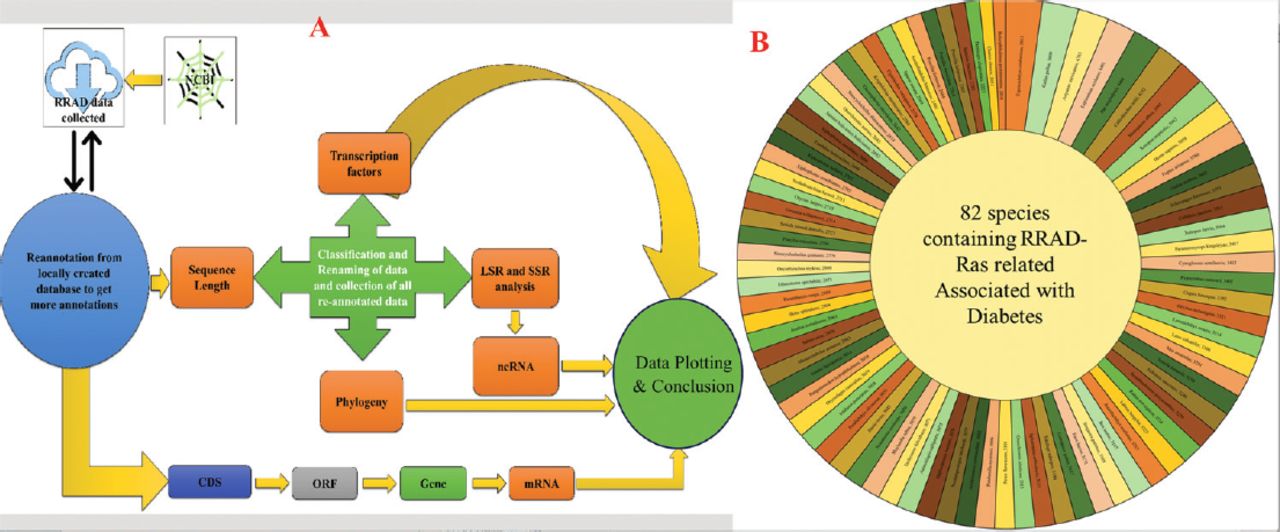
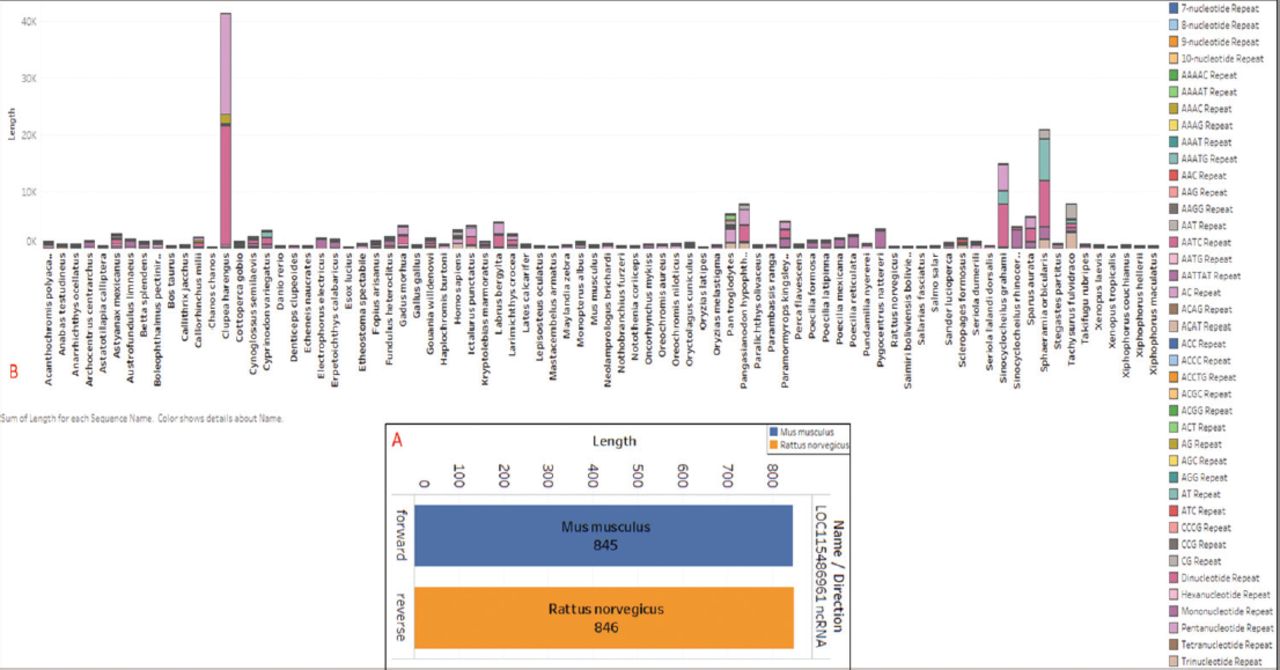
- Showing the A) graphical abstract of the methodology followed for the study; and B) sequence length of the Ras-related associated with diabetes (RRAD) genes.
The RRAD gene sequences were retrieved from the National Center for Biotechnology Information gene database. Using the search terms “Ras-related associated with diabetes” and “Ras-related glycolysis inhibitor and calcium channel regulator” with the Boolean operators “AND” and “OR”, 82 gene sequences from 82 different species were retrieved. The retrieved gene sequences were treated as the species name, and the sequence length was extracted to be plotted in sunburst form using Microsoft Excel (v.2018), as shown in Figure 1B. Furthermore, all genes were kept with AUGUSTUS (v.3.4.0), a bioinformatic tool used for predicting genes in eukaryotic genomes and reallocated to the ORF using Geneious Prime (v.2023.1, Biomatters Ltd.), a bioinformatic tool used for sequence analysis.26 The collected data were utilized to establish a local database, which was then employed for reannotation. This reannotation process involved utilizing the same database to gather additional annotations related to the RRAD gene. The collected data were used to create a local database for comparison and correlation with the online database for RRAD.
The function of repeat regions in cancer, including SSRs, is intricate and plays a diverse role in genomic dynamics. We emphasize the importance of targeting SSRs in the RRAD gene for potential cancer treatments. Repeat regions, known for their instability, can contribute to genetic mutations, potentially leading to the development of tumors. The SSRs, specifically, are linked to microsatellite instability observed in certain cancers. Integrating these insights into our broader understanding of cancer biology will provide a more nuanced perspective on the role of repeat regions in the complex landscape of cancer development and progression. Repeat regions were identified using 2 methods: the Phobos tandem repeat finder and the Geneious Prime’s mapper algorithm.27 For identifying polymorphic SSRs, the Phobos algorithm was used. The Geneious mapper algorithm was provided with the path of repeats within the genes, following the default parameter set with a minimum repeat length of 100 sequences and a mismatch tolerance of 0%. In this algorithm, repeats shorter than 10 nucleotides were disregarded. The Phobos tandem repeat finder was employed to carry out an exact and extended search for the repeat regions. Hidden repeats were subsequently removed, and the maximum size allowed for repeats was set to 10 nucleotides. Satellite sequences were limited to a maximum length of 100 base pairs. The letter “N” in the sequences was treated as a mismatch to ensure relevant details. Polymorphic SSRs were also identified using Phobos’s imperfect search algorithms. The repeat unit length ranged from a minimum of 2 base pairs to a maximum of 6 base pairs, and the imperfect search mode with typical analysis presets was utilized. Similar to the previous algorithm, “N” was considered a mismatch in the sequences.
The data were again processed for the further identification of transcriptional factors. The transcriptional factors were identified and extracted using the tfscan tool of The European Molecular Biology Open Software Suite (EMBOSS; v.6.5.7).28 The tfscan is a computational tool crucial in genomic analysis, aiding in the identification of transcription factor binding sites. Used in RRAD studies, tfscan enhances the understanding of regulatory elements. Its application provides valuable insights into the intricate regulatory networks that influence gene expression, a key aspect in cancer research. Instead of choosing a plant, insect, fungus, or vertebrate, we chose other organisms because we had the gene list from a diverse species. The sequence matching did not allow any mismatches, and a minimum length of 7 base pairs was selected for the transcriptional factors. We collected a total of 2 ncRNA from 2 species (namely, Rattus rattus and Mus musculus). These ncRNAs were further manually edited in the gene for clear visualization. All data were collected in one file, treated as a database, and reannotated to obtain more annotations within the RRAD genes.
Multiple Alignment using Fast Fourier Transform (MAFFT) is a powerful sequence alignment tool widely employed in bioinformatics. In this study, MAFFT likely played a pivotal role, enabling the alignment of sequences from different species. Its efficiency lies in its ability to handle large datasets swiftly, providing accurate alignments critical for understanding evolutionary relationships and identifying conserved regions within the RRAD gene. We aligned the data using the MAFFT aligner and plotted the tree using the Geneious Prime tree builder.29,30 Further, the tree file was exported and uploaded to the iTOL server (to better represent the tree with color effects), which was then plotted in a circular format using the same server.31
Results
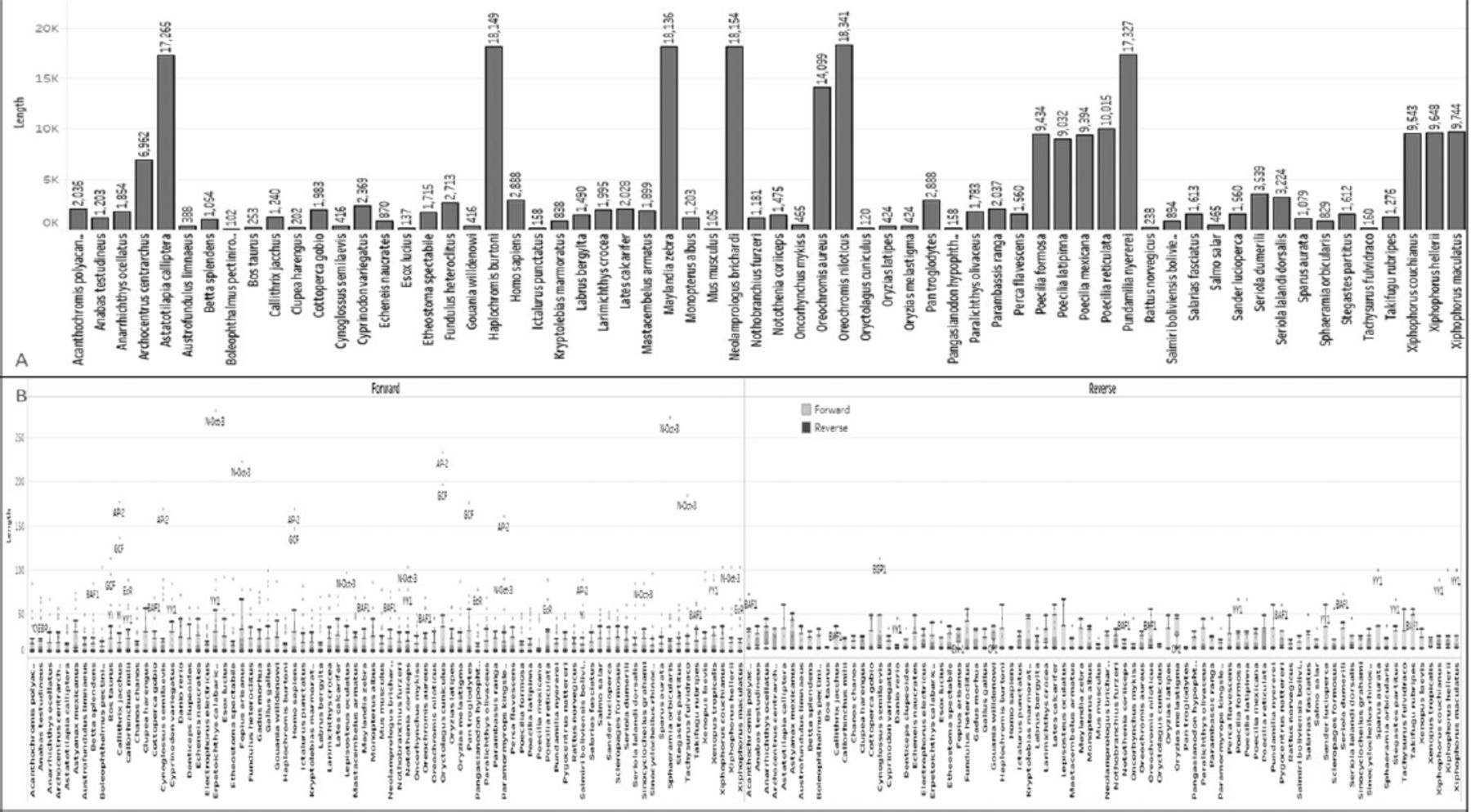
To aid in the decision-making process, the data were plotted using species names and sequence length to create a length graph. Among the 82 sequences analyzed, Erpetoichthys calabaricus exhibited the highest size, with a length of 5613 bp in the RRAD gene. On the other hand, Boleophthalmus pectinirostris had the smallest sequence length among all 82 sequences, measuring 2010 bp. Homo sapiens had a sequence length of 3858 bp, whereas Mus musculus had a sequence length of 3281 bp and Danio rerio had a sequence length of 3045 bp. Figure 1B provides further details on the sequence lengths of all 82 species.
The CDS of a gene refers to the segment of DNA or RNA responsible for encoding proteins. In the field of evolutionary biology, the acquisition of extensive data is crucial for understanding the various relationships among organisms. To gain comprehensive insights, we need to investigate several factors such as sequence length, nucleotide composition, expression patterns, splicing mechanisms, and structural features. These elements provide substantial information regarding the specific characteristics of organisms. Additionally, it is essential to compare the functions of the CDS with noncoding regions across different species and examine their temporal aspects. This comparative analysis can yield valuable insights into an organism’s biology.
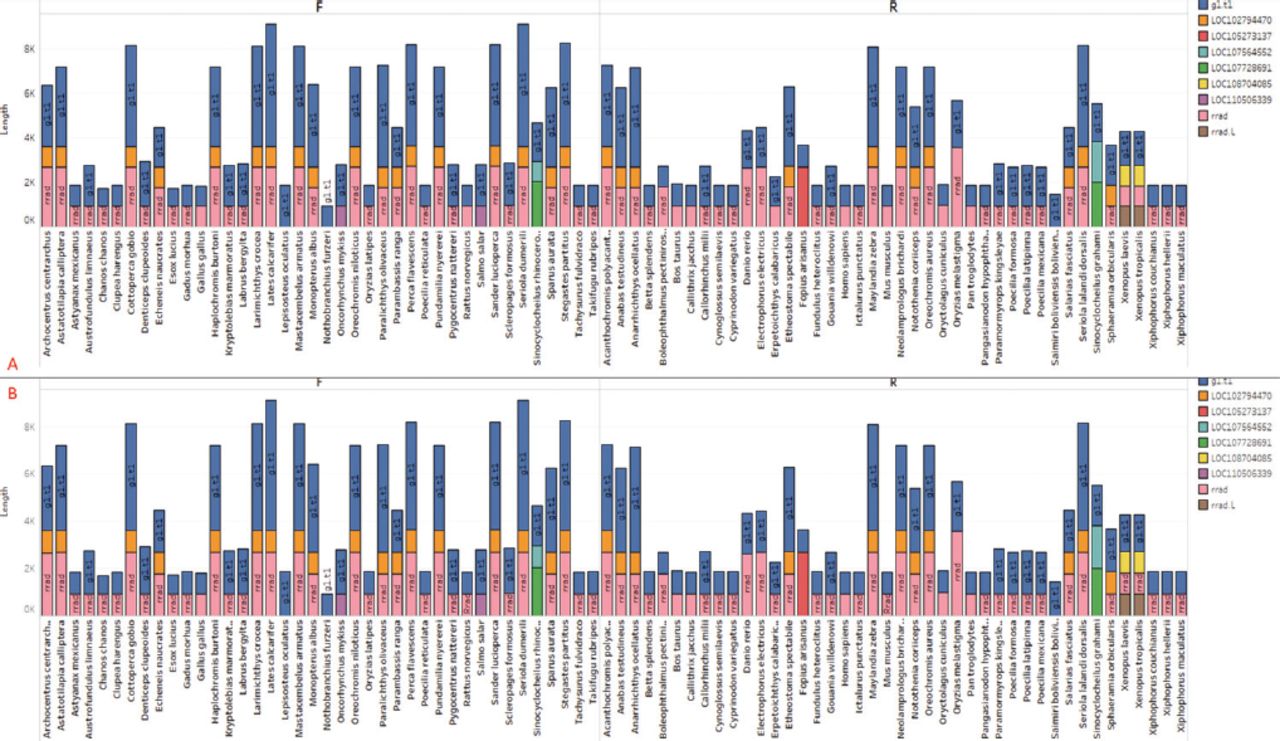
To provide a visual representation of these data, we plotted a detailed bar graph showing a total of 9 unique CDS types. Each CDS is assigned a different color and plotted alongside the organism’s name (Figure 2A). The local sequence database feature was utilized to reannotate the sequence, resulting in a collection of 368 unique CDS numbers from 82 RRAD genes or species. The analysis of these CDSs can greatly contribute to genome mapping and the development of gene therapy. Notably, Oryzias melastigma exhibits the longest CDS length in the RRAD sequence, ranging from 654-2861 bp. Conversely, Erpetoichthys calabaricus has the shortest CDS among all RRADs, measuring 516 bp. The human RRAD sequence contains a reverse form with a length of 927 bp. Gallus gallus has 2 CDSs with an identical sequence length of 912 bp, encompassing RRAD CDS and g1.t1 CDS. Mus musculus, in reverse form, has a size of 924 bp, including both CDSs (Figure 2A).

- Extracted A) CDS from all 82 species (9 types), plotted proportionally to the species and CDS types, with the sum of length for each organism broken down by direction. B) Extracted genes from all 82 species (9 types), plotted proportionally to the species and gene types, with the sum of length for each organism broken down by direction and color.
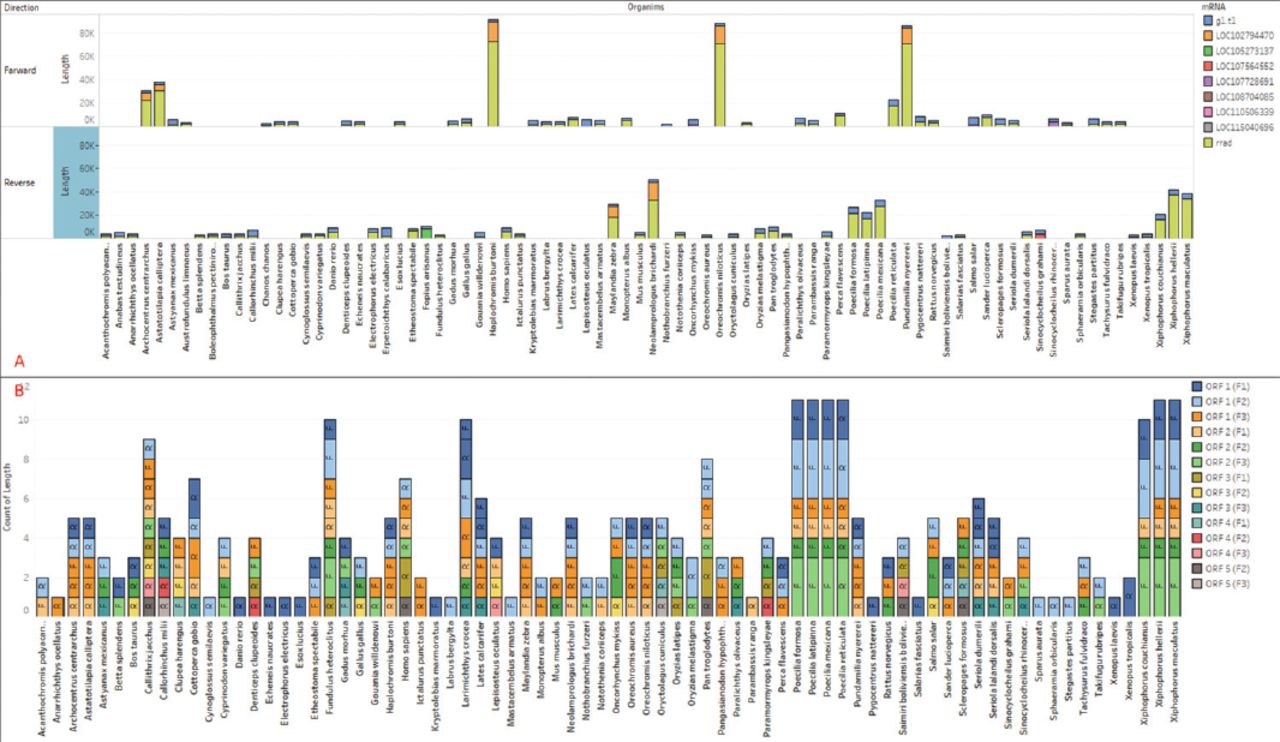
The CDS regions contain the genetic information that encodes proteins, which is vital for carrying out cellular functions. The mRNAs are single-stranded RNA molecules that complement one of the DNA strands in a gene. To gather insights into functionality, we extracted predicted genes from the CDS and mRNA sequences. The lengths of the extracted mRNAs and genes were plotted separately. In our analysis, we identified 9 unique genes in the sequence, and their lengths were visualized in relation to the corresponding organisms (Figure 2B). To facilitate accurate identification, the genes were classified using different colors. Notably, Homo sapiens’ RRAD gene exhibited 2 unique genes, one with a length of 3104 (g1) and another with a length of 3558 (RRAD), whereas Erpetoichthys calabaricus had the longest gene (RRAD) among all species of 5613bp. Furthermore, we plotted the mRNAs, which are the products of these genes (Figure 3A). The mRNAs were categorized based on their direction (forward or reverse) and are represented by different colors in Figure 3A.
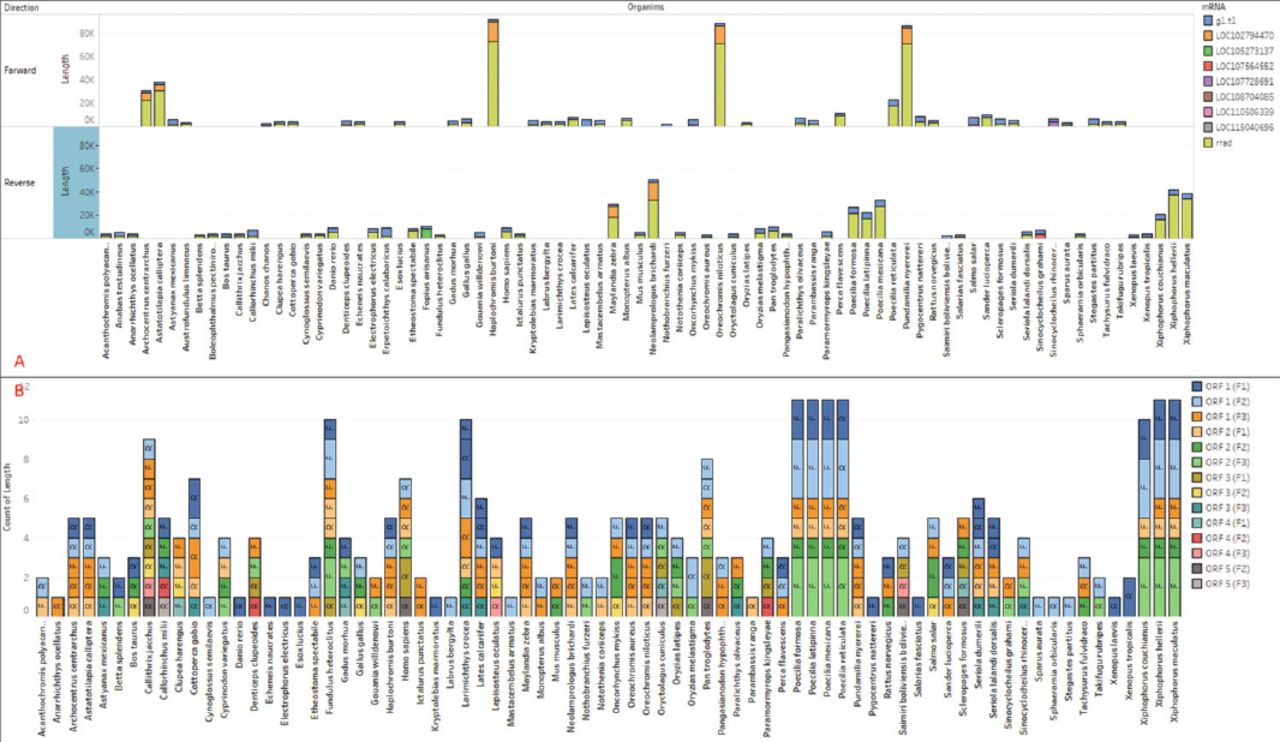
- Extracted A) mRNA from all 82 species, plotted proportionally to the species and mRNA types, with the sum of length for each organism broken down by direction in forward and reverse. B) Extracted ORF from all 82 species (14 types), plotted proportionally to the species and types, with the sum of length for each organism broken down by direction.
The ORF refers to a coding region within a DNA or RNA sequence that can be translated into a protein and does not contain any stop codons.32 Typically, an ORF begins with a start codon (AUG) and ends with a stop codon. A total of 14 types of ORF were identified based on their forward or reverse frame. The data extracted from the sequences of various species were plotted, showing the direction and count of ORFs in each region (Figure 3B). Among all species, Pan troglodytes has the longest ORF, measuring 852 bp (ORF1, frame2), while Oryzias melastigma has the smallest ORF, with a sequence of 392 bp (ORF1, frame2). In the RRAD sequence, Homo sapiens exhibits 7 ORFs. A total of 316 ORFs were collected from the sequences of all 82 species.
Additionally, ncRNA refers to functional RNA molecules transcribed from DNA that do not undergo protein translation. It includes various types such as miRNA, siRNA, piRNA, and ncRNA. These molecules regulate gene expression at the transcriptional and post-transcriptional levels. In the RRAD sequence analysis, we discovered that only 2 species contain ncRNA within the RRAD sequence. Mus musculus has a forward length of 845 bp, while Rattus norvegicus has a reverse length of 846 bp. To facilitate comparative understanding, the ncRNA for the 2 species is plotted in Figure 4A. Furthermore, all of the data were classified into forward and reverse forms, with “F” representing the forward direction and “R” denoting the reverse direction. The frames of the ORFs from each species are identified as F1, F2, and F3.
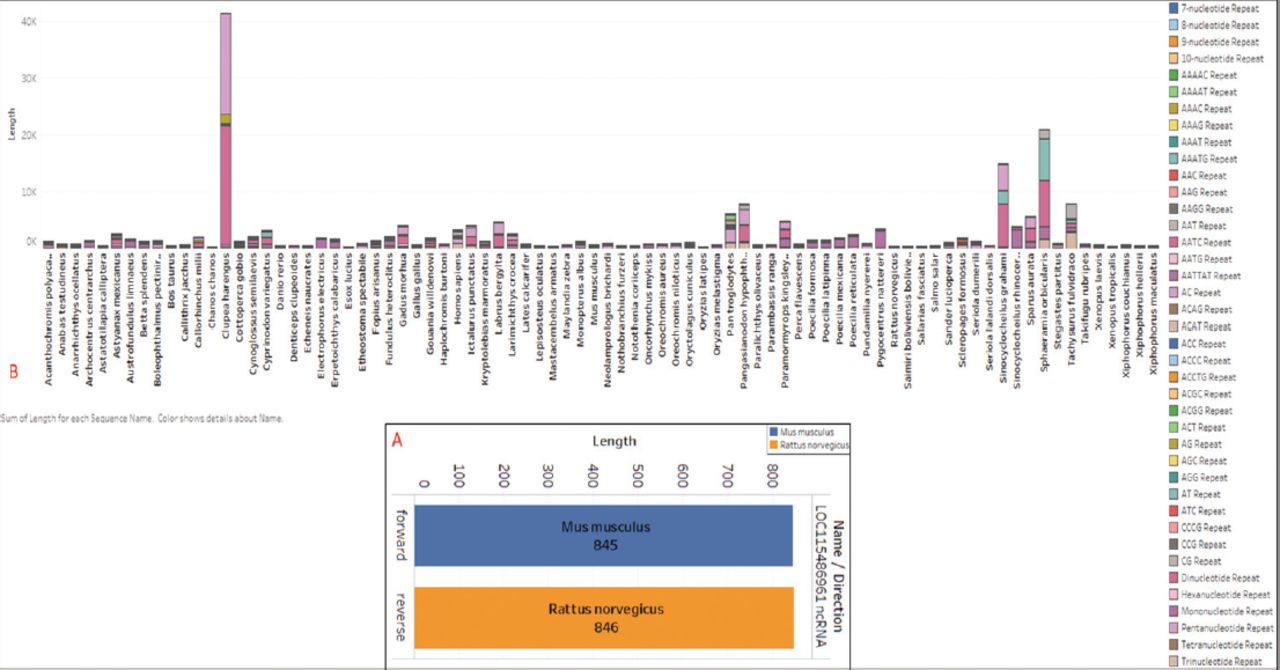
- Extracted A) ncRNA from all species, but only found in 2 species (Mus musculus and Rattus norvegicus) plotted proportionally to the species name and direction along with the length, with the sum of length for each order broken down by name. Color shows details on the organism. Both data were classified as forward and reverse by color. B) Extracted long-sequence repeats from all 82 species, plotted proportionally to the species, with the sum of length for each organism broken down by the species name.
Repeated sequences of repetitive elements are patterns of nucleic acids that appear in multiple copies throughout a sequence. These repetitive elements were initially identified due to their rapid re-association kinetics. The repetitive analysis was divided into 2 categories: long sequence repeats (LSRs) and SSRs. The LSRs were detected using the Geneious Repeat Finder algorithm, and were extracted and their cumulative count plotted per species (Figure 4B). Among all sequences, Neolamprologus brichardi exhibited the longest repeat. Figure 4B provides a comparative understanding of the data related to long-sequence repeats.
It is worth noting that tandem repeat sequences, particularly trinucleotide repeats, play a role in various human disease conditions. In our analysis, we utilized the Phobos algorithm to identify short-sequence and tandem repeats, and we plotted them collectively. We categorized the data into 43 types of SSRs for further analysis and assistance (Figure 5A). The species Sphaeramia orbicularis showed the highest count of dinucleotide repeats (n=93). The human RRAD sequence contains a total of 248 SSRs. These SSRs can serve as biomarkers and be utilized in various drug-related strategies.13,33,34 The dataset includes various types of trinucleotide repeats, dinucleotide repeats, mononucleotide repeats, 9-nucleotide repeats, and other SSRs, along with their corresponding sequences and positions.
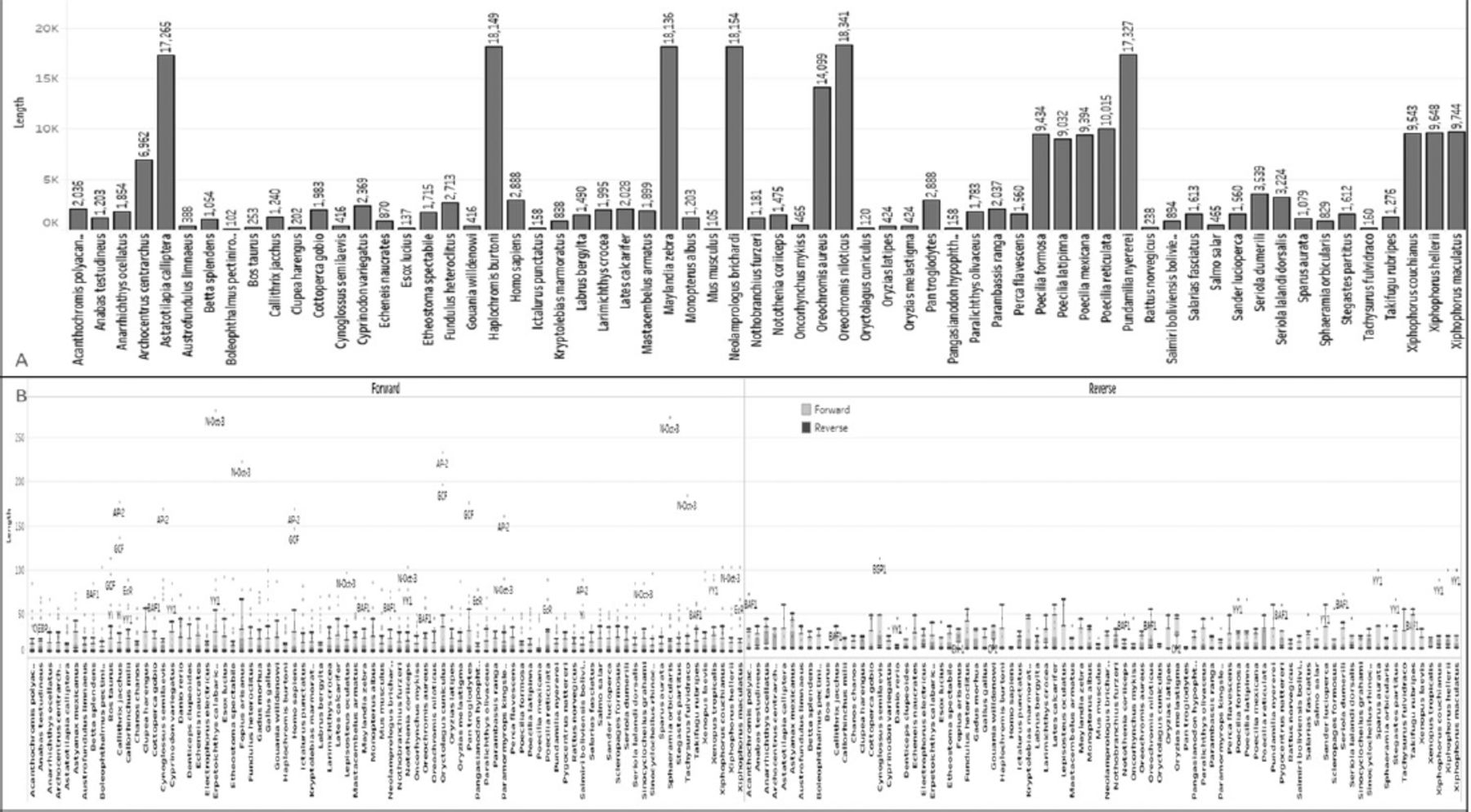
- Extracted A) short-sequence repeats from all 82 species (43 types of SSRs), plotted proportional to the species and types, with the sum of length for each organism. B) Extracted transcriptional factors from all 82 species, plotted in box and whisker graph proportional to the sum of length for each organism broken down by direction. Colour-coded according to forward and reverse transcriptional factors.
The TFs are sequence-specific DNA-binding factors, proteins that control the transcription rate by binding to a specific location in the DNA sequence. Specifically, TFs regulate genes to ensure they are expressed in the appropriate cell at the right time. A total of 6910 TFs from 82 RRAD sequences were identified in the transcriptional analysis using the tfscan tool, and plotted as a box and whisker chart. The length of the TFs ranges from 7-18 bp. The RRAD of Homo sapiens contains 116 TFs related to its sequences. Complete sequences were identified and classified based on their direction to ease data understanding and plotted using tableau. The dotted yellow circle was plotted forward, and the red process was plotted for the reverse transcriptional factors in the sequence (Figure 5B).
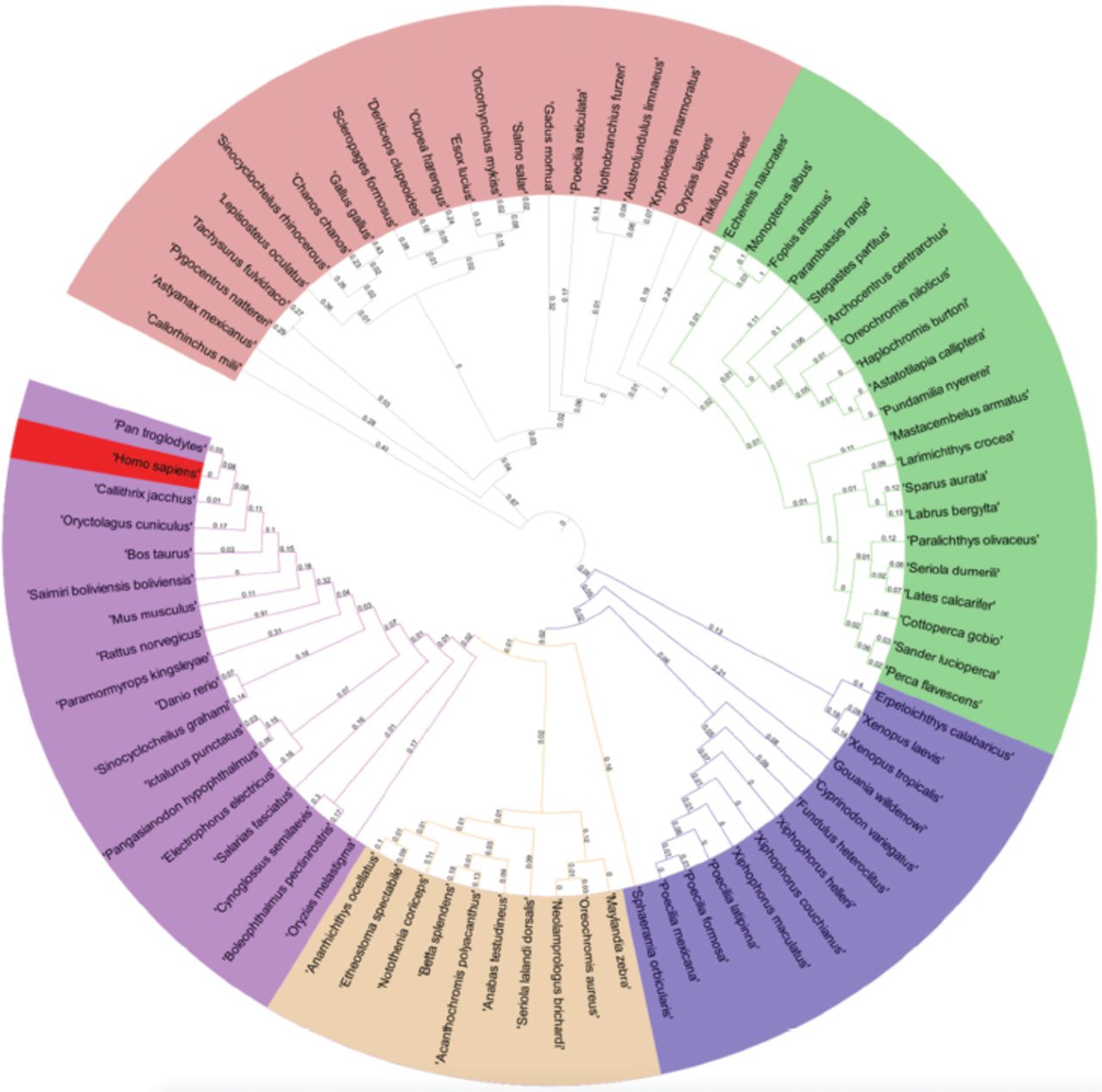
All species evolved from each other, and this process continues. The hypothetical diagram, which depicts a phylogenetic tree’s distances, can represent this relationship. The RRAD phylogenetic tree is the best method for showing and describing evolutionary relationships among organisms.35,36 We took all 82 species (sequences) for the phylogeny, aligned with the MAFFT aligner, and carried out the phylogeny using the Geneious tree builder. The phylogenetic tree was further modified in the iTol server to better visualize all species with the distance between them. The phylogenetic tree revealed that the RRAD of Homo sapiens is one of the youngest sequences, plotted in red. The RRAD of Callorhinchus milii seems to be one of the oldest sequences. Homo sapiens and Pan troglodytes are in the same clades and are similar to one another. Further, species such as Danio rerio and Oryzias melastigma are in the same outer clade as Homo sapiens, which seems nearer to others (Figure 6). Gallus gallus aligned in the pink region with Chanos chanos and in the outlier clade with Oryzias latipes.
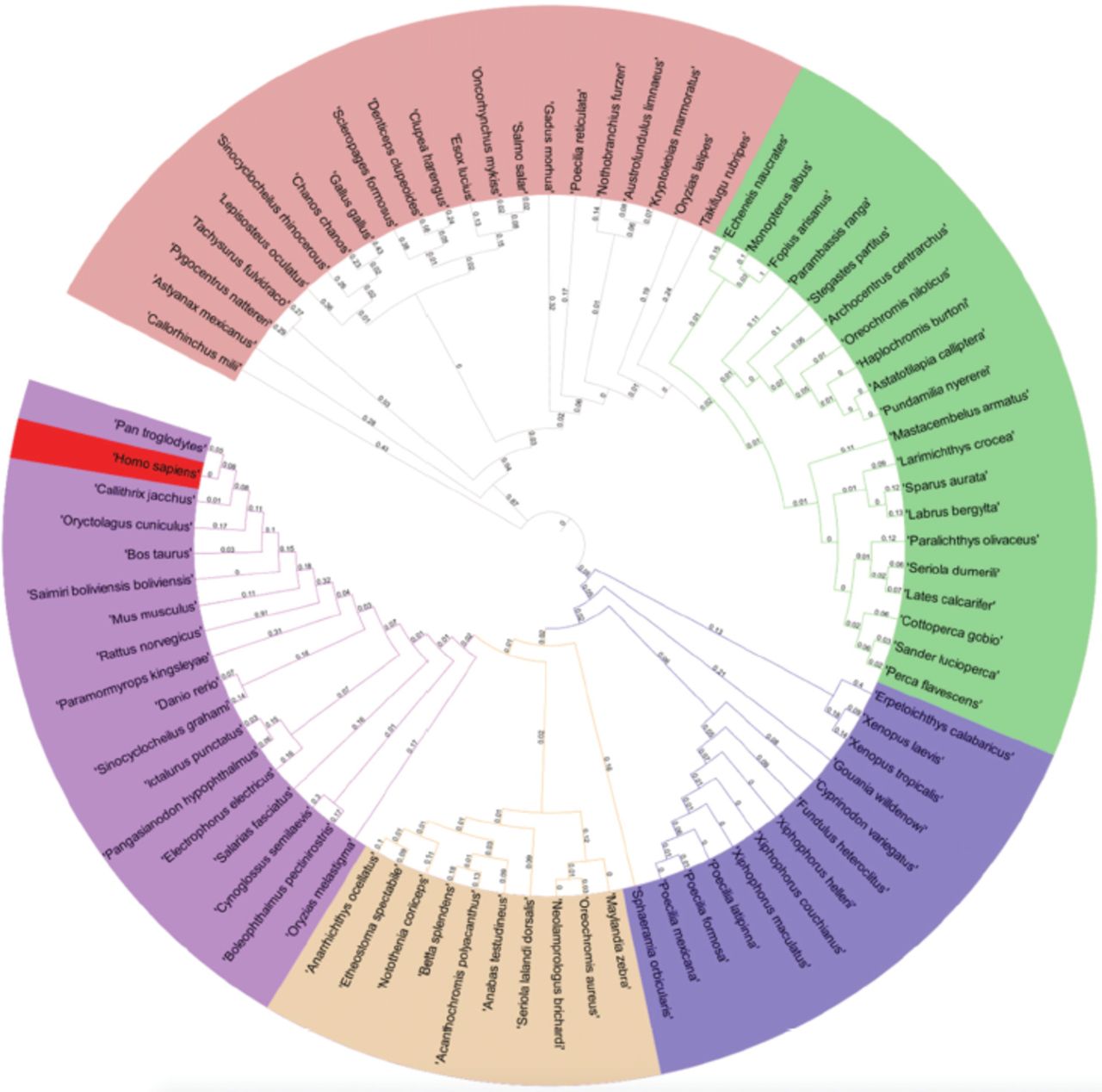
- Phylogenetic tree based upon the complete sequence of RRAD from all considered 82 species.
Discussion
It has been proposed that proliferating and cancer cells may prefer glycolysis to effectively prepare nutrients for rapid cell development.37,38 A study reported that a high-carbohydrate diet promotes Ras/Src-mediated transformation in Drosophila, and that transformed tumor tissues have diet-mediated insulin sensitivity, enhanced glucose absorption activity, and resistance to apoptosis.39 They also showed that acarbose and AD81 cause tumor cell death by inhibiting glucose uptake and Ras/Src AD81.39 According to Wang et al,4 RasV12-transformed T29H cells could take up more glucose due to RRAD downregulation, whereas the overexpression of RRAD inhibits glucose uptake and, hence, reduced glycolysis, which serves as a disadvantage for tumor growth.
In this study, the collected sequences related to RRAD from 82 different species were analyzed using tools and techniques such as Phobos, tfscan, EMBOSS, AUGUSTUS, and tableau. The analysis involved extensive data mining using Microsoft Excel to compare and examine the annotations and evolutionary aspects of RRAD sequences across different organisms. The current study is aimed at identifying distinct SSR, ncRNA, and CDSs that could be targeted while making cancer therapeutics to downregulate the RRAD gene in order to deprive the tumor or proliferating cells of glycolysis and decrease their proliferation.
Our in-depth analysis of all annotations and extracted data provides various exciting facts and strategies to inform further work on unveiling the specific site within the RRAD sequences for further expressional analysis in different organisms. Our extensive focused analysis of the different annotations provides a clear view of the sequences from different angles, which makes it easier to identify the region of the RRAD. The extraction of CDSs, genes, mRNA, and ORF gives a vast amount of information on their positions in different organisms, along with their length. The length comparison of all annotations also explains the number of genes (and locations) and other annotations within the genomes with base pairs. Further, we plotted all of the data using different graphical formats in the figures to allow for a more realistic understanding of the findings.
Identifying and targeting SSRs can be a good step towards breaching the genes of the RRAD in different organisms. It is better to break or add ligands to this coding region to upregulate or downregulate the expression. There could be many ideas and strategies for designing antisense mRNA or directly activating the immune system to produce the antisense mRNA. Targeting the SSRs or LSRs could be more beneficial. The identification of LSRs (>100bp) also plays a role in the different protein-producing genes and targets them specifically. In addition to the necessary annotations, we have identified the ORFs and ncRNA. We reannotated and predicted the ORFs using Geneious Prime to obtain more information on it. The ncRNA is only found in 2 species: Mus musculus and Rattus. Both ncRNAs were in different locations and different frames and directions. The TFs are located at different positions with different types in all species. A detailed analysis of the phylogenetic tree revealed that the RRAD of Homo sapiens is one of the youngest among all sequences (Figure 6). Callorhinchus milii seems to be one of the oldest among the sequences. Homo sapiens and Pan troglodytes are in the same clades and are very similar. Danio rerio and Oryzias melastigma are in the same outer clade as homo sapiens, which seems nearer than others (Figure 6). Gallus gallus aligned in the pink region with Chanos chanos and in the outlier clade with Oryzias latipes.
Study limitations
Our research provides important information on targeting RRAD for cancer treatment. However, we need to recognize certain limitations. The idea of inhibiting glycolysis is intriguing, but we should be cautious on assuming it universally applies to different organisms and cancer types.24,25 Relying on computer-based analyses with different tools could introduce biases. The suggested approaches, like targeting SSRs and ncRNA, require real-world testing to confirm their effectiveness and specificity. While our phylogenetic analysis is informative, it may not completely represent the evolutionary relationships. These factors emphasize the importance of carrying out thorough experiments to turn our computational discoveries into practical cancer therapies such as targeting SSRs and ncRNA, and their verification for efficacy and specificity in the context of HCC. Also, considering the complexity of cancer biology, cautious interpretation is advised, and comprehensive experimental validations are crucial before translating these computational insights into effective therapeutic interventions for HCC.
In conclusion, a large number of studies have already been published on RRAD, and many researchers are in the process of understanding its association with diabetes, cancer, and other diseases. Many have reported its relationship with cancer. Our study examined RRAD from 82 organisms from different families and provided a diverse ideological background to tackle the drug-designing biomarker identification problem. This study can be extended to identify a novel drug candidate from the identification of a particular region of the gene instead of complete knockdown planning, thus paving the way for sequence-based targeted and personalized medicine.
Acknowledgment
The author gratefully acknowledge the Deanship of Scientific Research at Shaqra University, Riyadh, Kingdom of Saudi Arabia, for supporting this work. The author also would like to thank Lola Mareno for English language editing.
Footnotes
Disclosure. Author has no conflict of interests, and the work was not supported or funded by any drug company.
- Received August 23, 2023.
- Accepted December 17, 2023.
- Copyright: © Saudi Medical Journal
This is an Open Access journal and articles published are distributed under the terms of the Creative Commons Attribution-NonCommercial License (CC BY-NC). Readers may copy, distribute, and display the work for non-commercial purposes with the proper citation of the original work.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.